SNP Genotyping Enhancements: From Traditional PCR to Double Helix Structural Oligonucleotides (STexS)
Introduction
As the Human Genome Project was accomplished, series of subtle variations of polymorphisms were reported within the population [1,2]. Among the various types, single nucleotide polymorphisms (SNPs) are known as the most abundant, with more than 9 million reports in the public databases [3,4]. In a molecular aspect, SNPs are nucleotide alterations at a specific region in the genome. While the term is applied to nucleotide replacements, SNPs can be broadened with biallelic variations such as insertions, deletions, and allele frequencies less than 1% [5,6]. SNPs generally occur around one per 1000 bp within the human genome [7]. However, the distribution is not even, as SNPs tend to cluster in noncoding regions more than several hundred folds compared to actual coding regions [8]. In hindsight, SNPs located in the noncoding region may be neglected. But various reports soon regarded both locales as an important region in disease developments and other genetic malfunctions [9,10]. As more SNPs were found to be an important overlap in certain conditions, it soon served as a crucial genetic or pharmacogenetic marker for future practices. The reason for such importance lies on which specific region, coding or noncoding, the SNPs are found. Even though noncoding regions do not contribute to protein coding, regulatory sites with SNPs can affect transcription rates, which alters the overall production of protein encoding. This process later triggers malfunctions that may develop disorders and various cancers [11-13].
As important the SNPs were known for molecular, pharmacogenetic, and cancer targets, so too did the importance to accurately depict and sort out targeted SNPs from normal, far more abundant gene strands. A common approach for this identification is a patient-control study involving large SNP genotypes in patient groups and healthy control populations to compare genotypes differences for all phenotypes under study. And the relationship between specific genotypes and phenotypes characterizes diseaserelated sensitive genes and is used to find encrypted proteins for disease prevention and treatment. Another major application in which SNP is used as an indicator is pharmacogenetic approaches [14-16]. Because the vast population size of the cohorts, discovering valuable SNPs that mark a specific disorder is rather trivial. However, the hurdle has been elevated as treatments shifted from prognostic to predictive medicine. While patients who already developed severely will inevitably have high portions of SNPs which indicates the disease, normal or soon to develop potential cohorts will have very low abundance which in some cases are not detected by previous methods [17,18]. Other reports indicate patients having false negative results in a certain detection test which results in failure to prevent further disease development or early treatments [19]. As technology developed, the method to efficiently detect SNPs also enhanced. The most common and efficiently known method is the utilization of Polymerase Chain Reaction (PCR).
Hence, the overall format regarding the use of PCR has not been replaced. The improvements were acquired by usually two ways; enhance the efficiency to estimate relative amplification rates or apply adjustment reagents to further boost the amplification rate of the targeted DNA template [20,21]. The method to elevate relative amplification rates was achieved through quantitative PCR (qPCR) [22]. Further enhancements to effectively sort out and specifically amplify targeted SNP templates were done by adding allelespecific primers, reagents which clamp unwanted DNA templates, or utilizing hydrolysis probes for melting curve analysis [23,24]. Many attempts to improve the detection rate of targeted SNPs bore fruit but was not complete. Because certain DNA strains would still attach the allele-specific primers and result in false positive misdiagnosis [25]. In order to achieve early detection of a target SNP while not binding with the normal DNA strand, novel approaches were needed. In this review, we address the methods of known SNP specific amplification methods and the recently introduced double helix structural oligonucleotide added PCR (STexS) [26].
Traditional SNP Genotyping Technologies
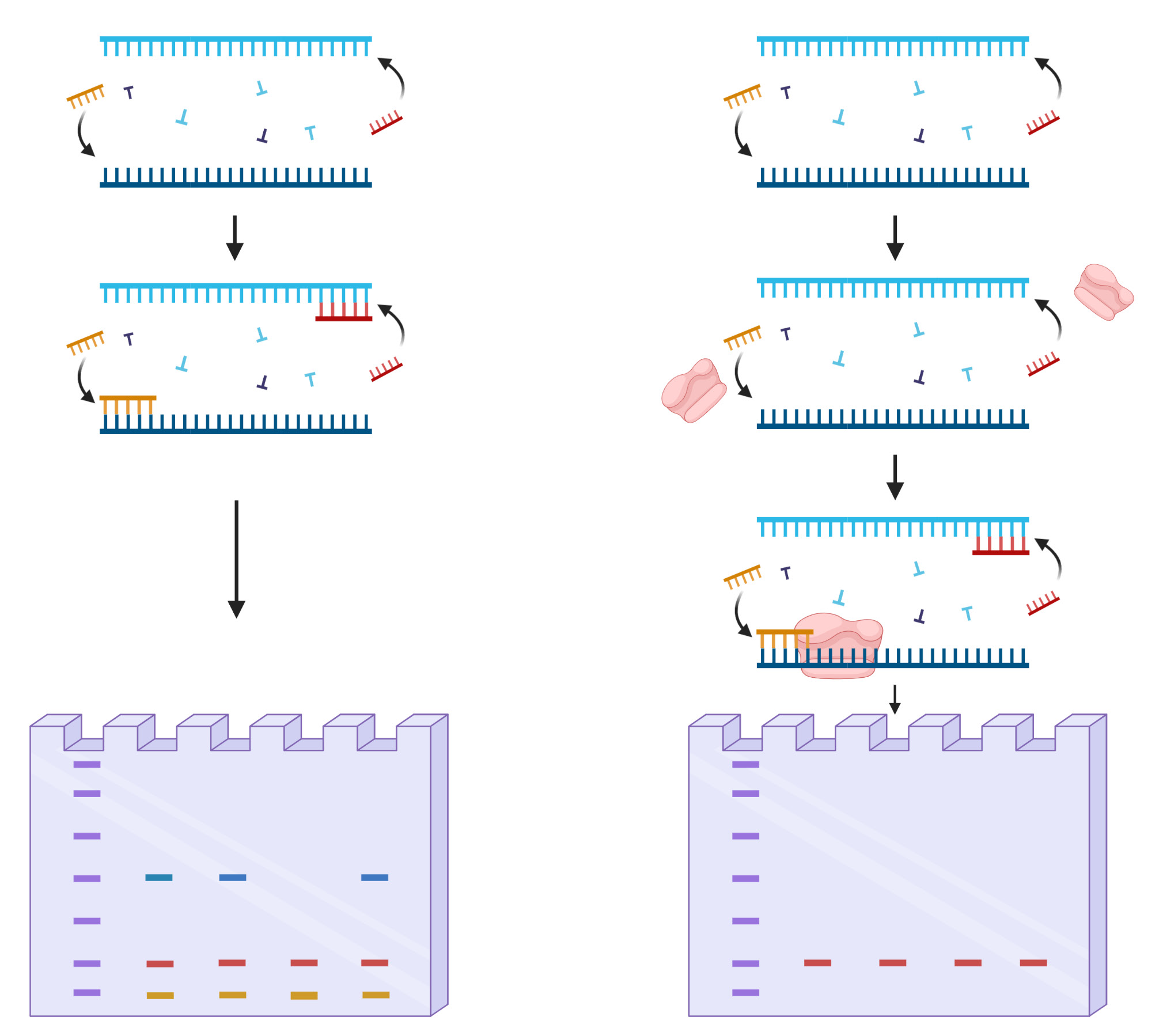
Figure 1: Basic schematics of the “HotStart” PCR.
SNP genotyping generally regards generations of allele-specific templates commercially or custom-made to increase amplification of wanted SNP-containing genes. The continuous efforts to manufacture efficient primers lead to many discoveries of genes which associates with a specific disorder such as Myeloperoxidase, N-acetyltransferase 1, and N-acetyltransferase which affects cancer susceptibility in Lung cancer, Bladder cancer, and colon cancer [27]. Other discoveries include SNPs located in CYP3A4 leading to prostate cancer and Thiopurine S-methyltransferase which SNPs alter the pharmacokinetic effects of anticancer drugs [28]. Although the roles of genes with certain SNP types were found, the method to effectively detect such allele in drastically low abundant patients were consistently held back due to limitations. The major limitation was nonspecific binds of normal DNA strands leading to primer template mismatches, resulting in false-positive detections to personnel who has the potential to develop the disorder. The “HotStart” PCR was a method to overcome such nonspecific amplifications by increasing temperature during the annealing process (Figure 1) [29]. However, SNPs consisting less than 1% in a clinical sample required something more sophisticated to discriminate normal DNA. The recently introduced STexS PCR platform combines the concept of “HotStart” PCR and allele-specific primers and further restrains nonspecific binds using a known method called amplification refractory mutation system-PCR (ARMS-PCR) [30]. ARMS-PCR is meant to detect any mutation involved single nucleotide changes including insertions and deletions [31]. This method greatly improves the efficiency to sort mismatched primers, but ARMS-PCR alone declines the amplification rate drastically below the limit of detection tampering the overall SNP genotyping.
The Implementation of Oligonucleotides
Oligonucleotides has been used alongside the standard PCR for various purposes including SNP detection [32,33]. Oligonucleotides are used as a probe to detect a specific sequence that are complementary to the oligonucleotides. Because it is mainly consisted of a relatively short 12~25 nucleotide, it is also referred as primers in a conventional PCR for detecting and multiplying wanted complementary gene targets. Oligonucleotides are majorly single stranded, thus when performing PCR, both forward and backward templates of a targeted template is required for maximum efficiency. However, in SNP detection, nonspecific bindings of mismatched gene strands lead to misleading results. While the process to completely block the large portion of normal genes to be amplified is a challenge, giving the targeted SNP strand enough cycles to amplify firsthand by delaying mismatched DNA copies will make enough difference to successfully perform SNP genotyping.
The STexS platform mentioned above uses a double helix forming oligonucleotide (discrimination boosting oligonucleotide, dbOligo) to fully utilize the concept of disrupting mismatched amplification. The annealing process is mainly contributed by the DNA polymerase (DNAP) that synthesizes matched or mismatched strands to a complete gene template, which in turn acts as an additional template for amplifying [34]. While intended matches of dbOligo does not boost or block the amplification rate compared to other commercially used primers, the kinetic energy of DNAP to attach and detach mismatched primers dwindles, resulting in a constant unstable state that ultimately delay the overall process of amplification (Figure 2). Combining with the “Hotstart” method, each dbOligo is deliberately designed to be at least 10 base pairs to adjust the annealing temperature to further block nonspecific bindings and primer dimers. Validation through previously reported SNPs contributing to several cancer types showed a reasonable contrast between matched and mismatched templates, further proving the importance of STexS method (Table 1).
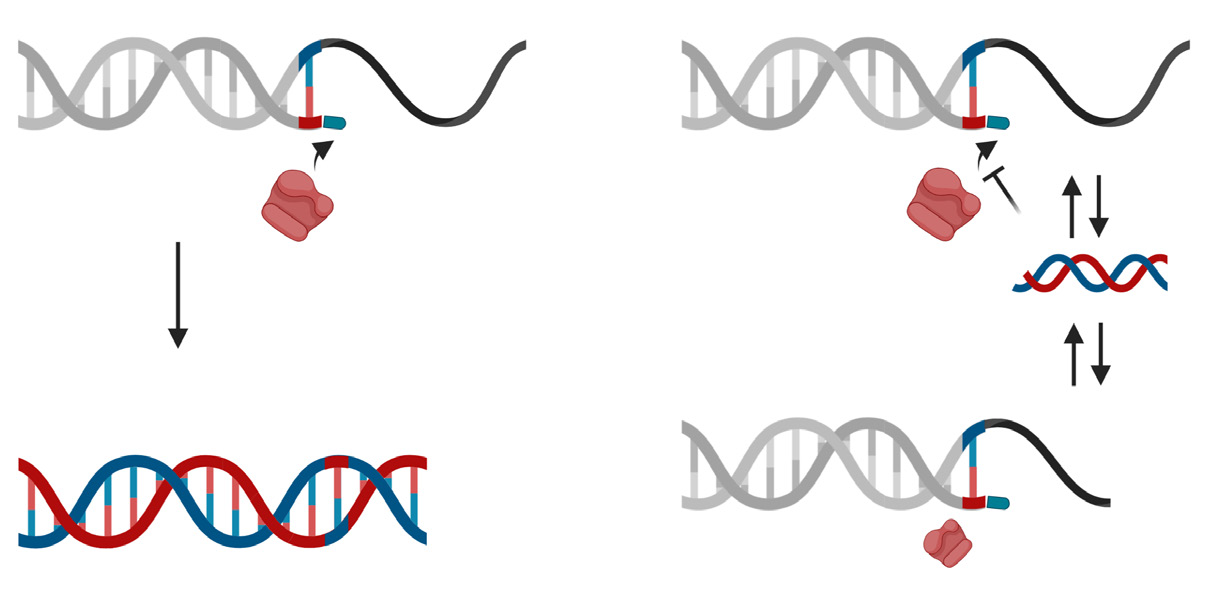
Figure 2: Difference of mismatched amplification between conventional PCR and STexS PCR.
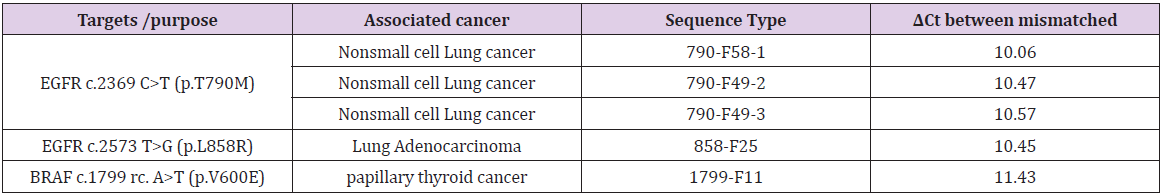
Table 1: SNP targets of different cancer and amplification difference between matched and mistmached DNA.
Conclusion and Future Directions
The ability to sort out even one mismatch between two different DNA strands is a fundamental technology not only limited to PCR techniques, but to other molecular extents. Fully discriminating matched targets from unwanted mismatched strands will also lead to improved specificity and sensitivity. Current diagnostics with various arrays and detection kits are challenged not just with its rate of success, but also constant incidents of false negatives misleading practitioners to judge poorly [35,36]. The importance of not neglecting cases of false negatives drastically increased, as contagious viral infections tend to spread among patients who cannot be scanned with conventional methods [37-39]. The recent spread of SARS-CoV-2 influenced generations of detection methods to effectively tackle and contain infections from further progression. However, due to the rapid mutation of the virus’s nature, the ongoing struggle seems to go ever on [40]. The STexS will definitely serve as a suitable bedrock for future detections arrays. Not just with currently known lethal mutated variants, the STexS platform can successfully detect hundreds of variants regarding both precedented and novel, hazardous and non-lethal but with potential to develop. Future applications for the STexS will be focused not only with SNPs related to early cancer detection, but also in precise diagnosis of the ongoing COVID-19. Further, the technology will improve molecular biology and medical science and reshape the current health care industry.



No comments:
Post a Comment
Note: Only a member of this blog may post a comment.