Childhood Hypopituitarism: A Three Decades of Experience from a Major Teaching Hospital in Central Region (Riyadh) Saudi Arabia
Abstract
Childhood hypopituitarism is a clinical syndrome of deficiency in
pituitary hormone production. Presentation varies from asymptomatic to
acute collapse depending on the etiology, rapidity of onset and
predominant hormone involved. A retrospective hospital-based cohort
study was conducted at the pediatric endocrine service, King Khalid
University Hospital Riyadh, Saudi Arabia (January 1989-December 2017).
All the patients (total of 202 patients) who were diagnosed to have
hypopituitarism, at the pediatric endocrine service, King Khalid
University Hospital Riyadh, Saudi Arabia (January 1989-December 2017). A
total of 202 patients were diagnosed to have hypopituitarism. Mean age
was 8-9years. Beside congenital causes, diversity of acquired causes
were encountered with a non-tumor causes being the commonest. Growth
Hormone Deficiency (GHD) was diagnosed in 152 (70.2%) patients. Isolated
GHD in 123 patients. Multiple pituitary hormone deficiency was the
diagnoses in 29 patients. Central adrenal insufficiency was present in
59(29.2%) patients. Diabetes insipidus associated with hypopituitarism
in 24(11.9%) patients. Childhood hypopituitarism is not that rare. High
index of suspension coupled with appropriate hormone assay, and head MRI
are essential for management.
Introduction
The pituitary gland is a midline structure located just beneath the
optic chiasm. It is composed of two main lobs, the predominant anterior
lobe which is known as adenohypophysis and the posterior lobe which is
known as neurohypophysis and the vestigial intermediate lobe [1].
Hypopituitarism is a clinical syndrome of deficiency in pituitary
hormone production. It might be a life threating condition. It can
result from disorders involving the pituitary gland, the hypothalamus or
the surrounding structures, such as tumor, inflammation, infection,
surgical destruction, radiation, traumatic or vascular insult. It might
be associated with birth trauma and perinatal asphyxia or midline
defects such as cleft lip, septo-optic hypoplasia and encephalocele.
Hypopituitarism could be partial or complete insufficiency of pituitary
hormone. Panhypopituitarism refer to involvement of two or more
pituitary hormone. However, involvement of one hormone only refer to
isolated or partial hypopituitarism. The younger the child is at the
time of presentation the more likely the etiology is to be congenital.
However, on occasions, congenital forms may present or get diagnosed
well after birth and conversely, some
children with acquired forms are discovered relatively early in life
[2-8]. This article reports on the clinical experience of childhood
hypopituitarism from a major teaching hospital, King Khalid University
Hospital (KKUH) in the central region of Saudi Arabia over more than 25
years, January 1989 to December 2017. KKUH is affiliated to King Saud
University and provides primary, secondary and tertiary health care
service to the local population and receives patients’ referral from all
over the country.
Materials and Methods
The medical records of patients who were diagnosed to have
hypopituitarism were retrospectively reviewed from January 1989 to
December 2017. Data included were age, sex, clinical presentation and
results of the relevant laboratory investigations and radiological
images. Magnetic Resonance Imaging (MRI) was done when appropriate. The
diagnosis of hypopituitarism was based on clinical suspicion suggested
by the appropriate hormonal testing. The various hormonal testing to
assess both the anterior and posterior pituitary gland functions, were
performed following the specific protocol [9]. Pituitary function was
evaluated after
1-2 months of any neurological procedure. Thyroid function was
periodically evaluated as to monitor for thyroid dysfunction.
Statistical Analysis
Descriptive data is provided. Dichotomous data is presented as
frequencies and percentage. Statistical Packages for Social Service
(SPSS version 21) was used for the statistical analysis of the data.
Results
During the period under review, January 1989 and December
2017, there were 202 patients with hypopituitarism, of these, 142
(70.3%) were males and 60 (29.7%) females. The mean age was 8-9
years (range 0-18 years). MRI brain was performed in 173 (85.6%)
of patients. The clinical presentation varied from asymptomatic
to symptomatic, the severity of which depends on the hormone
deficient and the cause of the disorder. Birth trauma, hypoglycemia,
neonatal hepatitis, optic nerve hypoplasia and other midline
defects where clues to the diagnosis. Beside congenital causes, a
diversity of acquired causes were encountered with a non-tumor
causes being the commonest. Those included Traumatic Brain
Injury (TBI), in three patients and Sub Arachnoid Hemorrhage
(SAH), in one patient, central nervous system infections were in
two patients, histocytosis in three patients, empty sella syndrome
in one patient and various tumors in the hypothalamic pituitary
region, the majority of which is craniopharyngioma, constitute 12
(5.9%) patients in our series.Growth Hormone Deficiency (GHD) was the commonest
deficient hormone among other hormones 152 (70.2%) patients.
Isolated Growth Hormone Deficiency (IGHD) 123 patients was more
common compared Multiple Pituitary Hormone Deficiency (MPHD)
in 29 patients (Table 1). Hypothalamic pituitary MRI abnormalities
were seen more commonly among cases with MPHD. Central
adrenal insufficiency was the second most common hormonal
deficiency seen in 59 (29.2%) (Table 2). Diabetes insipidus
associated with Panhypopituitarism seen in 24 (11.9%) patients
(Table 3). Hypothyroidism presented in 14 (6.99%) patients of
these five patients were isolated hormone central hypothyroidism,
while gonadotrophic hormone deficiency presented in 8 (3.9%)
patients.
Discussion
Hypopituitarism is not that rare in children, despite that there
is scant data where it is at best limited to case series. A study from
Spain documented an incidence and prevalence of hypopituitarism
to be 4.21 and 45.5 case per 100000 population respectively.
Hypopituitarism follows a smoldering course, unless it has an onset
with pituitary apoplexy, hence, more often it is likely to be missed.
Hypopituitarism is often associated with increased mortality
[10,11]. Magnetic Resonance Imaging (MRI) scan remains the
modality of choice for assessing the hypothalamic pituitary region
among patient with hypopituitarism. MRI scan precisely diagnose
abnormality of the adenohypophysis and neurohypophysis usually
the stalk. Normal pituitary MRI shows the anterior pituitary gland
as dark structure equal in intensity to gray matter on T1-weighted
imaging, while the posterior pituitary gland appears as a white
structure, “bright spot” (Figure 1). The “bright spot” correlates
well with the clinical presence of Diabetes Insipidus (DI) and it
is completely absent in some cases of congenital hypopituitarism
or it can be found ectopically located (Figure 2) in which case
DI is usually absent. Such radiological findings are sometimes
helpful in securing the congenital nature of hypopituitarism.
The presence of abnormality in the adenohypophysis and/or
neurohypophysis carries important prognostic factor. Patients with
such abnormalities usually develop MPHD compared to isolated
hormonal deficiency. A spectrum of MRI findings was observed
in our series ranging from normal to small or complete absent of
anterior or posterior pituitary together with absence or thin stalk
(Figure 3) [ 12].
The clinical presentation varies from asymptomatic to
acute collapse, depending on the etiology, rapidity of onset and
predominant hormones involved [2,5,13,14]. During neonatal
period and early infancy, hypoglycemia is considered the most
critical presenting feature and perhaps the most common feature
of congenital hypopituitarism. In some affected male, congenital
hypopituitarism can present at birth as isolated microphallus
therefore from isolated GH deficiency or combined GH and
gonadotropin deficiency. Additionally, noninfectious hepatitis can
be seen in some children with congenital hypopituitarism. This
is usually suspected if there is hepatomegaly and abnormal liver
enzymes predominantly indicating cholestasis and is confirmed
by the presence of characteristic giant-cell transformation of
hepatocytes on liver biopsy. The condition is usually self-limiting
and remits over the first few months of life without permanent liver
damage [15-25].
Septo-optic dysplasia, which also known as de Morsier
syndrome, is the most common cause of ‘’midline defect syndrome’’
in which congenital hypopituitarism is one component of this
disorder. In its complete form, this syndrome combines hypoplasia
or absence of optic chiasm, optic nerves or both, agenesis or
hypoplasia of the septum pellucidum, corpus callosum or both
(Figure 3) (the function of which is uncertain) in 50% of cases
(hence, the designation,”septo”) and hypothalamic insufficiency.
The underdevelopment of the optic nerves is associated with
variable degree of vision impairment ranging from mild loss to
complete blindness.

Various tumors in the hypothalamic pituitary region can lead
to hypopituitarism upon presentation. The majority of tumors
in our series were craniopharingioma, constitute 12 (5.9%).
Commonly they present with symptoms related to mass effect of
the tumor on the neighboring structures. However, other tumors
in the hypothalamic pituitary region can lead to hypopituitarism
such as glioma and astrocytoma. A child in our series presented
with recurrent headache and visual impairment at age 13 years
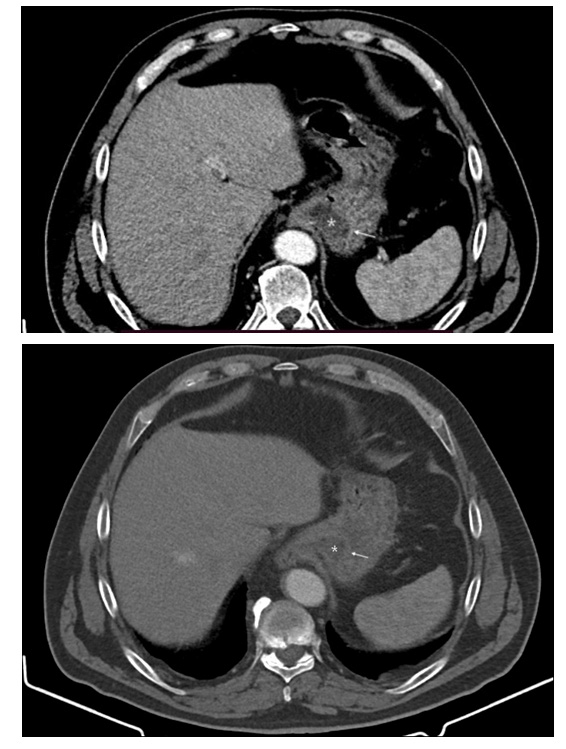
proved to have giant macroprolactinoma on MRI (Figure 4)
[26-33]. Such findings indicate the importance and the role of
pituitary and hypothalamus MRI scan in determining the cause.
Langerhans Cell Histocytosis (LCH) is a rare and heterogeneous
disease, characterized by accumulation and cloned proliferation of
immature dendritic cells in different organs, present in 3 (1.5%) of
patients [34,35].
Cancer survivors have a continuous risk for developing
hypopituitarism even if they had a normal initial pituitary function.
Surgical procedure to remove the tumor may lead to injury to
the pituitary gland and result into hypopituitarism. Additionally,
radiotherapy treatment for the tumors in the head and neck region
may cause damage to the pituitary gland. Different hypothalamic
pituitary axis has different sensitivity to radiation [36-40].
Traumatic Brain Injury (TBI) and Sub Arachnoid Hemorrhage
(SAH) are being more frequently reported as etiology for
hypopituitarism. The incidence of TBI 100-150 in 100,000. Posttraumatic
hypopituitarism is observed in 5.4 to 40 % of patient
with history of TBI usually presenting as isolated deficiency in
most cases. Hypopituitarism has been observed in 19% of patient
with ischemic stroke and 47% of patient with SAH presenting
as an isolated deficiency in most cases. This could be due to lack
of awareness [41-46]. Efforts need to be made to sensitize the
clinicians about the existence of hypopituitarism in such patients.
Langerhans Cell Histocytosis (LCH) is a rare and heterogeneous
disease, characterized by accumulation and cloned proliferation of
immature dendritic cells in different organs, present in 3 (1.5%) of
patients [35,35].
Central nervous system infections known to have high incidence
in developing countries and are known to cause hypopituitarism.
In this study hypopituitarism was observed in 2 patients (0.5%).
Both of these patients developed hypopituitarism secondary to
streptococcal infection [47-50]. Empty sella syndrome usually
associated with pituitary hypofunction. Like other cohorts it was
a rare diagnosis in our series [51-53]. Growth Hormone Deficiency
(GHD), the most common hormonal deficiency and present with
a wide spectrum of findings. The endocrine abnormality of GHD
manifest either as an isolated deficiency (IGHD) or it may be
present as Multiple Hormone Deficiency (MPHD) [54-56]. Central
adrenal insufficiency accounts for the second most commonly
encounters in this study, however, it is the most lethal. All except
one, had multiple anterior pituitary hormone deficiency. This was
present in 59 (29.3%) patients [57-59]. One patient passed away
at age 4 years from severe hypoglycemia during gastroenteritis
despite that she received hydrocortisone stress dose. She was found
seizing in her bed with unrecordable blood glucose. She developed
cerebral edema followed by DI and passed away because of brain
death. Central Diabetes Insipidus (CDI) or Anti-Diuretic Hormone
(ADH) deficiency may be associated with hypopituitarism, due to
impairment of posterior pituitary gland or hypothalamus in 24
(11.9%) patients. Patients presented with polyuria, polydipsia and
high serum sodium of more than or equal 150 mmol\l with diluted
urine [60-62]. Furthermore, congenital isolated hypogonadotrophin
is extremely rare in this study, 8 (3.9%) patients out of 202 patients,
similar to what had been reported in other cohorts [63-64].
Conclusion
This is the largest report of childhood hypopituitarism from
Saudi Arabia. High Index of Suspicion coupled with timely and
appropriate hormonal assay are essential for the management.
A head MRI scan is critical in determining the specific etiology.
It needs to be diagnosed and treated early to prevent associated
mortality and morbidity. The etiology differs in childhood, as nontumor
causes are common than in adults. Supervision and care by a
pediatric endocrinologist is crucial and when the child grows older
appropriate transition to an adult endocrinologist and experienced
reproductive endocrinologist.
Development of a Low Calorie Ready to Serve
Beverage from Hibiscus cannabinus L - https://biomedres01.blogspot.com/2020/03/development-of-low-calorie-ready-to.html
More BJSTR Articles : https://biomedres01.blogspot.com



No comments:
Post a Comment
Note: Only a member of this blog may post a comment.