Quantum Chemical, Molecular Docking, and ADMET Predictions of Ketorolac and its Modified Analogues
Introduction
In computer aided drug design system, molecular docking, nonbonding interactions, and ADMET predictions are important criteria to evaluate newly designed molecules [1]. A group of non-steroidal anti-inflammatory drugs (NSAIDs) has been routinely medicated for arthritis, fever, and pain through common mechanistic pathways of cyclooxygenase (COX) inhibition of prostaglandin synthesis [2]. More of, in the cell bio assay COX enzyme especially COX-2 enzyme catalyzes the inflammatory mediator prostaglandin synthesis, prostacyclin and thromboxane as a consequence, suppression of COX-2 enzyme activity could be of therapeutic usage [3], [4]. In vivo selective COX-2 inhibition strategy of Ketorolac drugs is well known for cancer treatments due to their structural diversity, high efficacy, low side effect over other NSAIDs drugs [4,5]. Apart from that, Ketorolac salt also has selective inhibition of DDX3 protein through intra and intermolecular H-bond formation in the diagnosis of several types of cancer [6].
By rational investigation and functionalization of NSAIDs drugs, researchers put forward their studies on more potential action with reduced side effect and multiple diseases such as cancer HIV, neuro-degenerative diseases [7,8]. In this research we scrutinized computationally various derivatives of Ketorolac and their mode of action with amino acids of respected protein in view of their structural properties and future implementation on drug discovery. NSAIDs drugs have some adverse effects depending on the type and nature of unusual physical condition of the body and on the limit of dose [9]. Recently, it has been seen the trait of modifying drugs using halogens and alkyl group play important role in improving drug performance. Drug modification is another alternative way to search better agent, which can increase the selective action of drug and reduce the side effect [10]. Herein, we report the optimization of some Ketorolac derivatives to investigate their biochemical behaviour on the basis of quantum mechanical approach.
The free energy, electronic energy, enthalpy, dipole moment, electrostatic potential, HOMO-LUMO gap, hardness, softness, and chemical potential have been calculated. Molecular docking and nonbonding calculation have been performed to understand the binding affinity, mode(s) and interaction between drugs and the amino acid residues of human prostaglandin synthase protein (5F19). All the derivatives show better thermal stability, and chemical reactivity and few of the derivatives have better binding affinity, nonbonding interactions. From the regarding quantum chemical studies, we are assuming that, some of the designed compounds may have profound effect as drug.
Methods and Materials
Computational Details
In computer aided drug design, quantum mechanical methods are widely used to predict thermal, molecular orbital, and molecular electrostatic potential properties [11]. Initial geometry of Ketorolac (K) was taken from the online structure database named Chem Spider [12]. Geometry optimization and further modification of all structures carried out using Gaussian 09 program [13]. Density functional theory (DFT) with Becke’s (B) [14] three-parameter hybrid model, Lee, Yang and Parr’s (LYP) correlation functional [15] under Pople’s 6-31g (d,p) basis set has been employed to optimize and elucidate their thermal and molecular orbital properties [16]. Initial optimization of all compounds was performed in the gas phase. Dipole moment, electronic energy, enthalpy, free energy, electrostatic potential and atomic partial charge are calculated for all the compounds.
Frontier molecular orbital features HOMO (highest occupied molecular orbital), LUMO (lowest unoccupied molecular orbital) were calculated at the same level of theory. For each of the drugs, HOMO-LUMO energy gap, hardness (η), softness (S), and chemical potential were calculated from the energies of frontier HOMO and LUMO as reported considering Parr and Pearson interpretation [17,18] of DFT and Koopmans theorem [19] on the correlation of ionization potential (I) and electron affinities (E) with HOMO and LUMO energy (𝜀). The following equations are used to calculate hardness (η), softness (S), and chemical potential (μ);
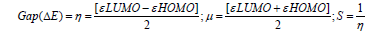
Molecular Docking and Nonbonding Interactions
Molecular docking simulation was performed to understand the mechanism of the prostaglandin H2 (PGH2) inhibition by newly designed Ketorolac analogues and their binding affinity and mode(s) with target protein [20]. The 3D structure of aspirin acetylated human cyclooxygenase-2 (PDB ID: 5F19) was obtained in pdb format from online protein data bank (PDB) database [21]. All hetero atoms and water molecules were eliminated using PyMol (version 1.3) software packages [22]. Energy minimization of the protein implemented by Swiss-Pdb viewer software (version 4.1.0) [23]. Than optimized drugs were subjected for molecular docking study against human prostaglandin synthase protein (5F19). Finally, molecular docking simulation was performed by PyRx software (version 0.8) [24] considering the protein as macromolecule and the drug as ligand. In this analysis, rigid docking was performed where, all rotatable bonds were converted into non-rotatable with the center grid box size 20.8612, 37.5501 and 59.3402 Å along x, y and z directions respectively. After docking, both the protein and ligand structures were saved in .pdbqt format required by Accelrys Discovery Studio (version 4.1) to analyze and visualize the docking result and search the interactions between ligands and target protein [25].
ADMET Predictions
In drug discovery, computational predictions are using to explore absorption, distribution, metabolism, excretion, and toxicity (ADMET) which saves on time and investment. AdmetSAR online database was utilized to predict ADMET properties of Ketorolac and its analogues [26].
Figure 1: Chemical structure of Ketorolac (K) and its designed analogues.

Result and Discussion
Thermochemical Analysis
Simple modifications of molecular structure significantly influence the structural properties including thermal and molecular orbital parameters. From the free energy, and enthalpy values, spontaneity of a reaction and stability of a product can be predict [27]. In drug design, hydrogen bond formation and nonbonded interactions also influenced by dipole moment. Increased dipole moment can improve the binding property [28]. From thermodynamic data (Table 1), the free energy of Ketorolac is -859.6743 Hartree, where K1 shows the highest negative value (-1319.2329 Hartree). The –Cl substitution (K1) influence the free energy significantly. Highly negative free energy suggesting stable the configuration. Again, the dipole moment of Ketorolac is 0.5665 Debye where K4 shows the maximum dipole moment (5.2017 Debye) due to substitution of –CN group (Figure 1).
Table 1:The stoichiometry, molecular weight, electronic energy, enthalpy, free energy in Hartree and dipole moment (Debye) of Ketorolac (K) and its derivatives.

Figure 2:Most stable optimized structures of Ketorolac and newly designed drugs by analogues. Optimized with B3LYP/6- 31g (d, p) level theory.

Molecular Orbital Properties
The HOMO-LUMO energies, hardness, softness, chemical potential of all drugs are presented in Table 2. The electronic absorption relates to the transition from the ground to the first excited state and mainly described by one electron excitation from HOMO to LUMO [29]. The chemical hardness, softness, and chemical potential values depend on the energy of HOMO-LUMO [8,30]. Kinetic stability increase with the increase of HOMO-LUMO gap. As a result, removal of electrons from ground state HOMO to excited state LUMO requires more energy. In our studies, Ketorolac shows the HOMO-LUMO gap 5.1935 eV, where K4 have lowest energy gap (4.1334 eV) and chemical potential (-7.4028 eV) with highest softness (0.4838 eV) which may contribute higher chemical reactivity (Figure 3).
Figure 3: Frontier molecular orbital (HOMO-LUMO) and related transition energy of Ketorolac (K) and K4.

Table 2: Energy (eV) of HOMO, LUMO, gap, hardness, softness and chemical potential of the designed drugs.

Molecular Electrostatic Potential Analysis
Molecular electrostatic potential (MEP) was calculated at B3LYP/6-31G (d) level of theory to forecast the reactive sites for electrophilic and nucleophilic attack of all optimized structures [31]. Red colour represent maximum negative area which favourable site for electrophilic attack, blue colour indicate the maximum positive area which favourable site for nucleophilic attack and green colour represent zero potential area. MEP displays molecular size, shape as well as positive, negative and neutral electrostatic potential regions simultaneously in terms of colour grading. It is seen from MEP map, region having the negative potential are over electronegative atom (oxygen atoms) and having positive potential are over hydrogen atoms. Here, the maximum positive potentiality is found for Ketorolac is -0.2475 a.u (deepest red) for oxygen atoms and the highest positive potentiality of K5 is +0.1837 a.u (deepest blue) of hydrogen atoms (Figure 4)
Figure 4: Molecular electrostatic potential maps of Ketorolac (K) and K5.

Molecular Docking and Nonbonding Interactions Analysis
Binding affinities and ligand-protein interactions are summarized in Table 3. Greater negative values of binding affinity indicate stronger binding between drugs and the receptor protein. Strong hydrogen bonding is the most significant contributing factor in increasing binding affinity of drugs with the receptor (Figure 5). Non-covalent interactions such as hydrogen bond, halogen bond and hydrophobic interaction are involved in the binding of examined drugs. Recently, it is reported that, hydrogen bond of < 2.3 Å are able to increase the binding affinity by several magnitude [32] and halogen bonds have almost similar importance as hydrogen bond in chemical and biological system [33]. The binding affinity of Ketorolac is -8.7 kcal/mol where, K2, K4 and K5 have considerably increased to -10.0, -9.5 and -9.1 kcal/mol respectively. Decreased binding affinities are found in case of K1 (-8.6 kcal/mol) and K3 (-8.5 kcal/mol). Significant hydrogen and halogen bonding observed in K5 (Figure 6), which not only contributes in increasing binding affinity but also increase binding speciality.
Figure 5:Docked conformation of K2 at inhibition bounding site of receptor protein (5F19).

Figure 6: (A) Nonbonding interactions of K5 with receptor protein 5F19. (B) Hydrogen bond surface of 5F19 with K5.

Table 3: Binding affinity and nonbonding interactions of Ketorolac (K) and its modified derivatives.

Here, H=Conventional hydrogen bond, C= Carbon hydrogen bond, A= Alkyl, Aps= Amide-pi stacked, PA= Pi-anion, PS= Pi-sigma, Pal= Pi-alkyl, PPS= Pi-pi stacked, Ppt= Pi-Pi T shaped, X= Halogen bond
Nonbonding interactions are important consideration of drug protein conjugation. Except conventional H-bond frequently observed non-bonding interactions are CH/π, OH/π, and CH/O. Upon introduction of -Cl group in K1 structure conventional H-bond predominated with both Cysteine and Asparagin moiety within closer distance which is very reasonable since of chlorine contribution to total conjugation. On the other hand, methoxy group promoted several H-bonds through structural similar amino acids such as Asparagin, Glutamine, and Ariginine also in this case CH/π observed with Cystein, Proline and Leucine part of the protein. A variety of binding sites including both conventional and non-conventional H-bonds observed upon fluorine substitution, in where Arginine, Aspartic acid positioned most favorable distance toward the drug. Apart from that, CH/π interactions observed for Cystein, Tyrosine and Proline. No such contribution of conventional H-bonds observed during -CN group substitution rather than π/ alkyl non-bonding interactions observed with multiple amino acids. Lastly, in the K5, short distance halogen bonds observed with Aspartic acid. Another important Pi-Pi T shaped (Ppt) interaction observed in K4 and K5.
ADMET Predictions
ADMET calculation has performed to investigate the safety level of designed analogues after administration in the human body. According to Admet SAR data (Table 4), Ketorolac shows II category acute oral toxicity and rest of the analogues show III category acute oral toxicity, which suggesting less toxicity of analogues than parent drug. All the analogues are non-carcinogenic, show positive response for blood brain barrier (BBB) and human intestinal absorption criteria. All drugs are P-glycoprotein non-inhibitor where, P-glycoprotein inhibition can interrupt the absorption, permeability and retention of the drugs [34]. However, all the compounds show weak inhibitory feature for human ether-a-gogo- related gene (hERG) which can lead to long QT syndrome [35], so furthermore study of this aspect is necessary.
Table 4: Selected pharmacokinetic parameters of Ketorolac and its designed derivatives.

Here, NI= Non inhibitor, WI= Weak inhibitor, NC= Non carcinogenic
Conclusion
From quantum chemical calculations, all the analogues are thermally and configurationally more stable than Ketorolac. Also have smaller HOMO-LUMO gap and higher chemical reactivity than parent drug. From docking simulation, K2, K4, and K5 show higher binding affinity than K, where most significant interactions are observed for K5-5F19 complex. ADMET results predict that, analogues are non-carcinogenic, and relatively less harmful for oral administration. Considering above investigation, K2, K4, and K5 can be potent new possible candidate for the better performance.



No comments:
Post a Comment
Note: Only a member of this blog may post a comment.