An Integrative Genomics Approach for Associating Genetic Susceptibility with the Tumor Immune Microenvironment in Triple Negative Breast Cancer
Introduction
Triple-negative breast cancer (TNBC) represents a diverse group of breast cancers that are characterized by lack of expression of the estrogen receptor (ER), progesterone receptor (PR) and lack of amplification of the human epidermal growth factor (HER2) [1- 2]. It is a heterogeneous disease representing 15–20 % of all new breast cancers diagnosed each year in the US women population and accounting for 25% of all breast cancer related deaths annually [3]. Well known risk factors include age, ethnicity and genetics. TNBC has high prevalence in younger premenopausal women, primarily African American women and BRCA1 mutation carriers [1]. The lack of ER and PR expression and the lack of HER-2 amplification has significantly reduced targeted treatment options for patients with TNBC [1]. Currently, cytotoxic chemotherapy remains the main therapeutic modality [1]. Data from many studies over the past two decades has shown significant benefit of chemotherapy in the neoadjuvant, adjuvant and metastatic settings [4]. However, the benefits of systematic chemotherapy may be offset by the high risk of recurrence and poor clinical outcomes [2,4]. It is estimated that fewer than 30% of women with metastatic TNBC survive 5 years after diagnosis, and virtually all women with metastatic TNBC will ultimately die of the disease [4]. In addition, the benefits of chemotherapy are offset by the side effects which impair the quality of life for many patients [4]. Thus, there is an urgent need for the discovery of molecular makers and targets for the development of novel less toxic targeted therapeutics and precision prevention strategies.
Immunotherapy is revolutionizing the management of multiple solid tumors and offers new opportunities for the treatment of TNBC [5-7]. TNBC has the greatest incidence of patients with a robust tumor immune infiltrate [6], and therefore is amenable to immunotherapy. Currently, many immunomodulating agents for TNBC are undergoing clinical trials [6-8]. Potential TNBC immunotherapies currently under development include tumor vaccines, immune checkpoint inhibitors, antagonists of immune suppressive molecules and adoptive cell therapies [9-10]. Multiple promising clinical trials of anticancer vaccines and immune checkpoint inhibitors are ongoing [5-7]. Preclinical data has indicated that α-lactalbumin has the potential as a vaccine target for inducing safe and effective primary immuno-prevention as well as immunotherapy against TNBC [11]. However, the association between genetic susceptibility and tumor immune microenvironment has not been characterized. Understanding this relationship could have significant impact in identifying women at high risk of developing TNBC who could be targeted for vaccination as well as development of precision-prevention strategies for this aggressive and often lethal form of breast cancer.
Advances in high-throughput genotyping and reduction in genotyping costs have enabled discovery of genetic susceptibility variants and genes associated with an increased risk of developing breast cancer including TNBC using genome-wide association studies (GWAS) [12-16]. However, information on genetic susceptibility variants and genes have not been maximally leveraged and integrated with gene expression data to infer the association between genetic susceptibility and the tumor immune microenvironment. The objective of this exploratory study was to investigate the association between genetic susceptibility and tumor immune microenvironment in the basal –like subtype of TNBC. Our working hypotheses are:
a) Genomic alterations in genes containing genetic variants associated with an increased risk of developing breast cancer could lead to measurable changes associating genetic susceptibility with the tumor immune microenvironment; and
b) Genomic alterations in immune modulated genes affect entire molecular networks and biological pathways. For clarity, throughout this study we have considered single nucleotide polymorphisms (SNPs) associated with an increased risk of developing breast cancer as the genetic susceptibility variants, and the genes they map to as the units of association. Therefore, rather than focusing on individual genetic variants, this study focuses on genes, molecular networks and the biological pathways to understand the broader biological context in which genetic susceptibility variants operate in the tumor immune microenvironment in TNBC.
Material and Methods
Germline Mutations and Associated Genes
We have previously published a comprehensive catalogue of genetic variants primarily single nucleotide polymorphisms (herein referred to as genetic susceptibility variants) associated with an increased risk of developing breast cancer [12]. Details about methods used in the collection of genetic variants and genes from GWAS have been described in our earlier publication [12] which follow the guidelines proposed by the Human Genome Epidemiology Network for systematic review of genetic associations [17-21]. Here, we provide a brief, but detailed description of the data used in this study as well as the inclusion and exclusion criteria used. We reviewed published reports on GWAS on breast cancer. The reports were screened by title, abstract, and full-text review to identify the studies meeting our eligibility criteria. The studies were eligible if they met the following criteria:
a) Must have been based on a case-control study design using unrelated individuals,
b) Publications must have been of full length and published in peer-reviewed journals or online in English language,
c) Breast cancer must have been diagnosed by histological examination,
d) The study must have provided sufficient information such that genotype frequencies for both breast cancer and controls can be discerned without ambiguity, and
e) The studies must have used the appropriate and recommended statistical methods to infer the associations by taking into account the covariates and accounting for population structure and genetic background [17-18]. Studies with insufficient or incomplete information, reviews, studies reporting only intergenic regions, and studies with very small sample sizes were excluded from the study.
We manually extracted the information from the studies meeting our eligibility criteria and the accompanying Web sites containing supplementary data. The extracted information included SNP identification number (rs-ID); evidence of association as determined by the GWAS P-value; a composite of strong (P⩽10−7), moderate (P=10−5–10−6) and weak (P=10−2–10−4) association; gene name; and associated chromosome positions to which the genes map as determined by the dbSNP database [22] and the Human Genome Nomenclature database [23]. The literature search was cross referenced and validates using the GWAS catalogue Database [24]. The search yielded about 232 genes containing genetic variants associated with an increased risk of developing breast cancer used in this study. A complete list of genetic variants and genes associated with an increased risk of developing breast cancer along with sources or published reports from which they were derived is presented in Table SA provided as supplementary data to this report. Note that the entries (rows) in Table SA are more than the 232 genes containing genetic variants associated with an increased risk of developing breast cancer evaluated because of the same genetic variants and genes being reported or replicated in multiple independent studies, and alias names for the same genes.
Gene Expression Data
We used publicly available gene expression data on Caucasian women from the Gene Expression Omnibus (GEO) accession number GSE76124 on Caucasian women diagnosed with TNBC generated at Baylor University [25]. The experimental procedures have been fully described by the data originators [25]. The data set consisted of 54 patients diagnosed with the basal-like immune-activated and 60 patients diagnosed with basal-like immune-suppressed TNBC subtype. Informing this choice is that the basal-like subtypes constitutes 70 - 80% of TNBC and has the highest immune infiltrate [2, 26]. For controls, we used publicly available gene expression data on 100 cancer-free Caucasian women generated at Moffitt Comprehensive Cancer Center [27]. The control data set was downloaded from the Gene Expression Omnibus (GEO) database accession number GSE10780 [27]. The experimental procedures and methods of sample processing have been fully described by the data originators [27]. Both data sets were generated using the Affymetrix platform using the Human Gene Chip U133Plus 2.0 which contains (54,675 probe sets). Expression values were calculated using the robust multi-array average (RMA) algorithm as implemented in the Affymetrix platform. All the expression values ware on a log scale (log2).
Data Analysis
We performed whole genome analysis comparing gene expression levels between patients diagnosed with BLKIA and controls and between patients diagnosed with BLKIS and controls using a permutation t-test as implemented in Pomello and Gene pattern Software packages [28-29]. This unbiased approach was designed to identify genes containing genetic variants as well as genes without genetic variants. The rationale is that because GWAS discoveries explain only a small proportion of the phenotypic variation, limiting the analysis to genes containing genetic variants associated with an increased risk of developing breast cancer could miss important genes and pathways, as some of the genes containing genetic variants may be regulated by genes without genetic variants. Due to the relatively small sample sizes for each subtype, we did not partition the data into test and validation sets, as such an approach would lead to bias resulting from sampling errors. Instead we used the leave-one-out cross-validation procedure as our prediction and validation model to identify genes with predictive power. This approach has been used successfully in gene expression data analysis to eliminate bias [30]. We used the false discovery rate (FDR) procedure to correct for multiple hypothesis testing [31]. Genes were ranked based on the p-values and the FDR, and highly significantly differentially expressed genes were selected from each comparison. After ranking genes based on P-values and FDR, the genes containing genetic variants associated with an increased risk of developing breast cancer were identified by their names and corresponding gene symbols. Subsequently, we compared gene expression levels between patients diagnosed with BLKIA and patients diagnosed with BLKIS.
To determine whether the genes containing genetic susceptibility variants are co-regulated and have similar patterns of expression profiles, we performed unsupervised analysis using hierarchical clustering. We used the Pearson correlation coefficient as the measure of distance between pairs of genes and complete linkage as the clustering method. For each subtype, a set of differentially expressed genes between cases and controls was subjected to hierarchical clustering. This analysis was performed for GWAS genes only and for the combined set of GWAS and nonGWAS genes for each subtype. Prior to clustering, gene expression data was normalized using the median normalization, standardized and centered [32]. Hierarchical clustering was performed using Gene pattern [29].
To identify immune modulated molecular networks and biological pathways enriched for genetic susceptibility variants, we performed network and pathway analysis separately for BLKIA and BLKIS using the Ingenuity Pathway Analysis (IPA) software (http:// www.ingenuity.com) [33] For each subtype of TNBC evaluated, significantly differentially expressed genes were mapped onto networks and canonical pathways using the pathway build and design modules as implemented in IPA. The probability scores and the log P-values were calculated to assess the likelihood and reliability of correctly assigning the genes to the correct networks, functional relationships and biological pathways. We corrected for multiple hypothesis testing using the FDR procedure implemented in IPA. The molecular networks and biological pathways were ranked based on z-scores and log p-values; respectively. Gene ontology (GO) [34] analysis as implemented in IPA was performed to characterize the molecular functions, biological processes and cellular components in which the discovered immune modulated genes are involved.
Results
Associating Genetic Susceptibility with Immune Activation and Suppression
We performed analysis combining information on genetic variants and genes associated with an increased risk of developing breast cancer with gene expression data to investigate the association between genetic susceptibility and tumor immune microenvironment. We sought to discover and characterize gene signatures associating genetic susceptibility with BLKIA and BLKIS subtypes of TNBC. Table 1 presents a list of genes containing genetic variants associated with an increased risk of developing breast cancer as well as their estimates of GWAS and expression p-values for the two immunologic subtypes of TNBC investigated. Also presented in the table are the genetic variants reported to be associated with an increased risk of developing breast cancer based on original reports as documented in our previous report [12] and presented in the updated supplementary Table SA in this report. Comparison of gene expression levels between patients diagnosed with BLKIA and control samples revealed a signature of 198 significantly (P<0.05) differentially expressed genes containing genetic susceptibility variants and 119 highly significantly (P<10-6) differentially expressed genes without genetic variants. Comparison of gene expression levels between patients diagnosed with BLKIS and the controls produced a signature of 209 significantly (P<0.05) differentially expression genes containing genetic susceptibility variants and 119 genes (P<10-6) without genetic variants. Complete lists of significantly differentially expressed genes in BLKIA and BLKIS are presented in supplementary Table S1 for genes containing genetic variants and Table S2 for genes without genetic variants.
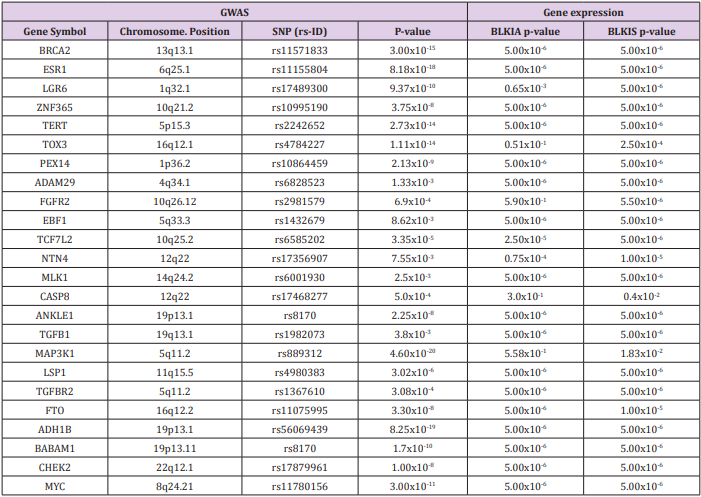
Table 1: List of genes containing genetic variants associated with the tumor immune microenvironment in patients diagnosed with the BLKIA and BLKIS subtypes of TNBC.2
The discovered immune modulated signature included the genes TERT, ESR1, CASP8, TOX3, ANKLE1, LGR6, PEX14, ADAM29, EBF1, TCF7L2, NTN4, BRCA1, BRCA2, LSP1, TGFB1, FTO and MLK1 which have been directly associated with an increased risk of developing TNBC [12]. The discovered immune modulated signatures also included the genes TOX3, LSP1, MAP3K1 and TGFB1 associated with an increased risk of developing both TNBC and non-TNBC [12]. The remainder of the genes contained genetic variants associated with unspecified type of breast cancer. This was expected because majority of GWAS have not been breast cancer type-specific [12]. Interestingly, among the genes containing genetic variants associated with immune modulation in the BLKIA and BLKIS subtypes included the genes BRCA1, BRCA2, TP53, PTEN, STK11 and CDH1 containing mutations with highpenetrance, genes ATM, BRIP1, CHEK2, PLB2, BARD1, NBN and RD50 harboring mutations with moderate penetrance and the genes MAP3K1, FGFR2, LSP1, TNRC19 which harbor mutations with low penetrance [35]. Genes containing genetic variants with weak to moderate association were also associated with BLKIA and BLKIS. Overall, there was significant overlap in differential gene expression levels between genes associated with BLKIA and those associated with BLKIS (Table 1).
To address the hypothesis that gene expression may differ between BLKIA and BLKIS and to identify a gene signature distinguishing the two tumor immune microenvironments, we performed additional analysis comparing gene expression levels between BLKIA and BLKIS. This analysis produced a signature of 60 genes containing genetic susceptibility variants distinguishing patients between the two tumor immune microenvironments.
Patterns of gene expression profiles in BLKIA and BLKIS
Immune modulation is a complex process and likely involves many genes acting in concert to shape the tumor immune microenvironment. To address the hypothesis that genes containing genetic variants associated with an increased risk of developing breast cancer are co-regulated and have similar patterns of expression profiles in BLKIA, we performed unsupervised analysis using hierarchical clustering as described in the data analysis subsection. First, we performed hierarchical clustering using only the 198 genes containing genetic variants associated with an increased risk of developing breast cancer associated with BLKIA. Subsequently, we performed hierarchical clustering combing the 198 genes containing genetic variants with the 118 genes without genetic variants. Here we sought to investigate whether genes containing genetic susceptibility variants are co-regulated and have similar patterns of expression profiles with genes without genetic variants. The results showing patterns of expression profiles for the 198 gene signature containing genetic susceptibility variants are presented in Figure 1. The results showing patterns of expression profiles based on the combined set of genes are presented in Figure 2. The investigation revealed that genes containing genetic variants associated with an increased risk of developing breast cancer are coregulated and have similar patterns of expression profiles (Figure 1). Additionally, the investigation revealed that genes containing genetic variants associated with an increased risk of developing breast cancer were co-regulated and had similar patterns of expression profiles with genes without genetic variants (Figure 2). Genes containing genetic variants reported to be directly associated with TNBC were co-regulated and had similar patterns of expression profiles with other genes. Interestingly, genes containing genetic variants with weak to moderate associations were co-regulated and had similar patterns of expression profiles with genes containing genetic variants strongly associated with Breast cancer. To address the hypothesis that genes containing genetic variants associated with an increased risk of developing breast cancer are co-regulated and have similar patterns of expression profiles in BLKIS, we performed hierarchical clustering as described in the data analysis subsection. Likewise, first we performed hierarchical clustering using only the 209 genes containing genetic variants associated with an increased risk of developing breast cancer associated with BLKIS. Subsequently, we performed hierarchical clustering combining the 209 genes containing genetic variants with the 119 genes without genetic variants.
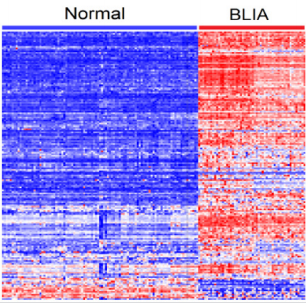
Figure 1: Patterns of gene expression profiles for the 198 genes containing genetic susceptibility variants significantly associated with tumor immune microenvironment in the 54 patients with BLKIA and the 100 control samples. Columns represents patients and rows represent genes. The red and blue colors indicate upregulation and down regulation; respectively.
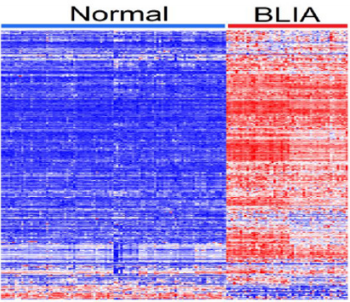
Figure 2: Patterns of gene expression profiles from combined analysis of the 198 genes containing genetic susceptibility variants and the 118 genes without genetic variants in patients with BLKIA. The columns represents patients and rows represent genes. The red and blue colors indicate upregulation and down regulation; respectively.
The results showing patterns of gene expression profiles for the 209 genes containing genetic variants are presented in Figure 3. The results showing patterns of expression profiles based on combined analysis are presented in Figure 4. The investigation revealed that genes containing genetic variants associated with an increased risk of developing breast cancer are co-regulated and have similar patterns of expression profiles (Figure 3). Similarly, genes containing genetic variants associated with an increased risk of developing breast cancer were co-regulated and had similar patterns of expression profiles with genes without genetic variants (Figure 4). Genes containing genetic variants with weak to moderate associations were co-regulated and had similar patterns of expression profiles with genes containing genetic variants strongly associated with Breast cancer. This is a significant finding considering that the genetic variants identify from GWAS have small effects but acting together could have large effects. in both BLKIA and BLKIS subtypes of TNBC, genes containing genetic variants associated with an increased risk of breast cancer were co-regulated and exhibited similar patterns of expression profiles. The similarity in patterns of gene expression profiles among genes containing genetic variants associated with an increased risk of breast cancer suggests that the genetic variants in genes of similar biological functions are likely to act together rather than individually in shaping the TNBC phenotype.
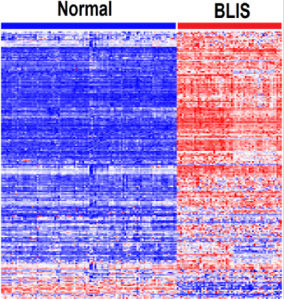
Figure 3: Patterns of gene expression profiles for the 209 genes containing genetic susceptibility variants and significantly associated with BLKIS. Columns represent patients and rows represent genes. The red and blue colors indicate upregulation and down regulation respectively.
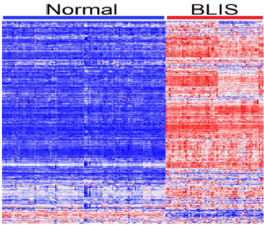
Figure 4: Patterns of gene expression profiles for the 209 genes containing genetic susceptibility variants and 119 genes without genetic variants for the BLKIS. The columns represent patients and rows represent genes. The red and blue colors indicate upregulation and down regulation respectively.
Molecular Networks and Biological Pathways
To discover and characterize the molecular networks and biological pathways modulated by the immune system, we performed network and pathway analysis for BLKIA and BLKIS separately. These analyses were performed using a combined set of genes. Here the goal was to gain insights about the broader biological context in which the genetic variants are likely to operate in the tumor immune microenvironments of patients diagnosed with BLKIA and BLKIS. Each analysis revealed gene regulatory networks containing genes with overlapping functions confirming our hypothesis. Figure 5 presents the results of network analysis for the BLKIA. The networks with the highest scores included:
a) Cancer with a z-score of 37. This network contained genes predicted to be involved in DNA replication, recombination and repair.
b) Cell cycle with a z-score of 31. This network contained genes predicted to be involved in DNA replication, recombination and repair.
c) Gene expression and cell cycle with a z-score of 26. This network contained genes involved in cell cycle and cancer. There was significant overlap in the molecular functions, biological process and cellular components in which the genes in the networks are involved.
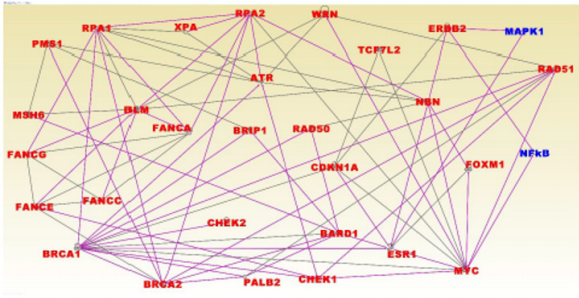
Figure 5: Molecular networks for genes associated with BLKIA subtype. The gene symbols in red font represent genes containing SNPs associated with an increased risk of developing cancer, non GWAS genes shown on blue font. The vertices or lines indicate functional relationships, while the color of lines simply indicates that the genes have overlapping functions.
The discovered networks included the genes BRCA1, BRCA2, PALB2, CHEK1, CHEK2, RAD50, BLM, MSH6 and MYC which have been directly associated with TNBC [12]. Among the genes in the networks were BRCA1 and BRCA2 containing mutations with highpenetrance, ATM, BRIP1, CHEK2, PALB2, BARD1, NBN and RD50 harboring mutations with moderate penetrance and the genes MAP3K1, FGFR2, LSP1, TNRC19 which harbor mutations with low penetrance [35] (Figure 5). Pathway analysis revealed immunemediated pathways including the role of BRCA1 in DNA damage response (P=1.10x10-25), Hereditary breast cancer (P=2.18x10-21), aryl hydrocarbon receptor (P=1.16x10-15) and the molecular mechanisms of cancer (P=7.74x10-12) signaling pathways, all which involved in TNBC. The results of network analysis for the BLKIS are presented in Figure 6. The networks with the highest scores included genes predicted to be involved in cellular development, cellular growth, proliferation and cell cycle (Z-score =52); DNA replication, recombination and repair, and cancer (z-score =35), and DNA replication, recombination and repair, cell cycle andcancer (z-score =24). Interestingly, among the discovered networks included the genes BRCA1, BRCA2, PALB2, RAD50, BLM, MSH6 and MYC containing genetic variants directly associated with and increased risk of developing TNBC [12]. Among the genes in the networks, the genes BRCA1 and BRCA2 contain mutations with high-penetrance, whereas the genes PALB2, BARD1 and RD50 harbor mutations with moderate penetrance [35] (Figure 6). Pathway analysis revealed immune-mediated pathways. The most highly significant pathways were: The role of BRCA1 in DNA damage response (P=6.69x10-27), the hereditary breast cancer (P=8.45x10-25), Aryl hydrocarbon receptor (P=7.85x10-19) and the molecular mechanisms of cancer (P=1.06x10-14) signaling pathways. (Figure 7) shows the role of BRCA1 in DNA damage response signaling pathway enriched for genetic susceptibility variants and the regulatory mechanisms involved in the pathway. The pathway revealed complex regulatory mechanisms involved in the tumor immune microenvironment. Among the regulatory mechanisms in the pathway included regulation of DNA damage and repair, regulation of gene expression as well as checkpoint regulation and checkpoint control (Figure 7). Among the genes found to be involved in DNA damage and repair (Figure 7) were: BRCA1 and BRCA2 containing mutations with high penetrance and directly associated with an increased risk of developing TNBC [12] and the genes ATM, CHEK1, CHEK2, and RD50 harboring mutations with moderate penetrance.
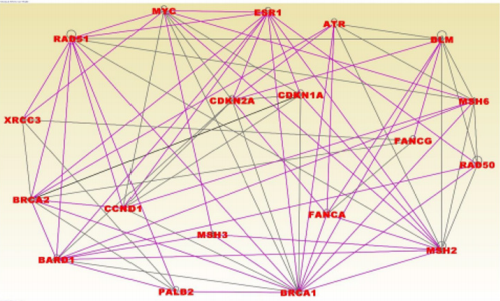
Figure 6: Molecular networks for genes associated with immune suppression in the basal-like (BLKIS) subtype. The gene symbols in red font represent genes containing SNPs associated with an increased risk of developing cancer, non GWAS genes shown on blue font. The vertices or lines indicate functional relationships.
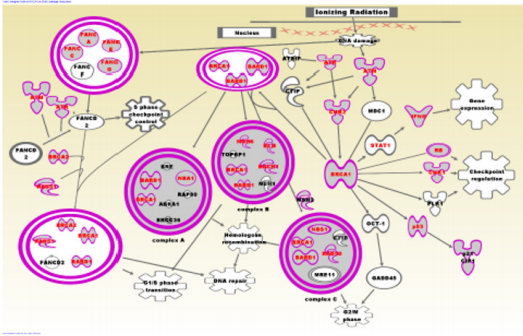
Figure 7: Role of BRCA1 in DNA damage response signaling pathway dysregulated in patients diagnosed with BLKIS. The gene symbols in red font represent genes containing SNPs associated with an increased risk of developing cancer. Genes without genetic variants are shown in black font. The solid lines with arrows indicate the direction of regulation. The double rings indicate regulatory complexes.
Discussion
Early detection and treatment offer the best chance of saving lives from TNBC. There is growing interest in exploiting intrinsic mechanisms of the host immune system in the treatment of TNBC [36]. With the discovery of antigens specifically expressed in TNBC cells and the developing technology of monoclonal antibodies, chimeric antigen receptors and cancer vaccines, immunotherapy is emerging as a novel promising option for TNBC [36]. In the genomics era, realizing immunotherapy requires understanding the association between the tumor immune microenvironment and genetic susceptibility. Currently, there is a knowledge gap between genetic susceptibility and the tumor microenvironment. This exploratory study was conducted to begin to address that knowledge gap by integrating information on genetic susceptibility variants and genes associated with an increased risk of developing breast cancer with gene expression derived from patients diagnosed with BLKIA and BLKIS TNBC. The investigation revealed immune modulated molecular signatures, gene regulatory networks and biological pathways enriched for genetic variants associated with an increased risk of developing breast cancer. Recently, comprehensive immunologic portraits of TNBC were reported using transcription profiling [37].
The main differences between that study and ours is that our study focuses on linking genetic susceptibility with the tumor immune microenvironment. To our knowledge, this is the first study to attempt linking genetic susceptibility with the tumor immune microenvironment in TNBC. The novel aspect of this study is that it demonstrates that integrative genomics leveraged with bioinformatics is a powerful approach to infer the association between genetic susceptibility and the tumor immune microenvironment. The clinical significance of this study is that by identifying women with compromised immune system who are at high genetic risk of developing TNBC, they could be prioritized for targeted vaccinations [36,38] and development of other precision prevention strategies. Likewise, the discovered gene signatures could be used as prognostic markers to stratify patients to guide treatment decisions at the point of care and realization of the precision medicine paradigm. The prognostic value of tumorinfiltrating lymphocytes in TNBC has been reported [39-42] as well as their correlation with chemotherapy [43-45]. Because early detection and early treatment offer a better chance of saving lives from TNBC, the link between genetic susceptibility and the tumor immune microenvironment could potentially result in paradigm shift in our knowledge of events initiating TNBC, which may also be relevant to understanding pathogenesis related to compromise in the immune system during tumorigenesis.
In this study, we focused on patients diagnosed with the BLKIA and BLKIS TNBC. Informing this choice were three critical points:
a) Within TNBC, the Basal-like disease predominates (70- 80%) and, from a biological perspective, should be considered a cancer-type by itself [1,46].
b) unlike other subtypes of TNBC, the basal-like has been clearly defined based on whether the immune system is activated (BLIA) or immunosuppressed (BLIS), with the worst prognoses being associated with BLIS tumors [25,46] and
c) the basal-like has the highest concentration of tumor immune infiltrates [47-48]. Importantly, in a recent literature search of twenty-five published studies comprising 22,964 patients, Mao Yan and colleagues were able to conclude that TILs had prognostic values only in TNBC for disease free survival (DSF) and Overall Survival (OS) [47-48].
The discovery of molecular networks and biological pathways containing BRCA1, BRCA2 and other genes involved in the DNA damage and repair is of particular interest. TNBC disproportionately affects BRCA1 mutation carriers [1]. The discovery of genes coregulated and interacting with BRCA 1 and BRCA2 genes suggests that other genes may be acting together with these genes. For example, pathway analysis in this study (Figure 7) revealed that BRCA1 gene encodes proteins critical for maintaining DNA integrity and genomic stability. This is consistent with reports in the published literature [49]. BRCA1 mutations cause homologous recombination (HR)-mediated DNA repair deficiency, genomic instability, and DNA-damaging agent hypersensitivity [49]. The discovery of BRCA1 and BRCA2 genes in the same networks and biological pathways, suggests that the two genes may have some shared functions in cancer predisposition, therapy response and immune modulation. However, the interactions of the two genes with other genes in the network and pathways suggests that there are also key differences between them indicating divergent roles for each protein.
We discovered immune modulated pathways in both the BLKIA and BLKIS. The clinical significance of these findings is that such pathways could serve as therapeutic targets. This is particularly important because the immune subsystem plays duo roles of suppressing tumor growth and promoting tumor growth in cancer patients [50], a phenomenon which explains the overlap in both differential expression and molecular functions of the genes discovered in this study. The clinical significance of these findings is that, while inhibition of immune response may result from an immunosuppressive state in the tumor microenvironment as observed in the BLKIS in this study and in a previous study [50], activation of the immune response may be triggered by an immune activation state of the tumor microenvironment as observed in the BLKIA in this report, which is consistent with previous reports [51- 52]. Such actions could make immune suppressive and immune activation pathways enriched for genetic susceptibility variants discovered here potential drug targets.
While our study suggests the association between genetic susceptibility and the tumor immune microenvironment, there is need to understand crosstalk between the tumor cell and the immune cells as well as the biological mechanisms regulating them, an aspect which is beyond the scope of this study, but a line of research worth pursuing in future. Importantly, although we did not attempt to study the mechanism by genetic susceptibility variants may directly regulate the tumor immune microenvironment, assessment of genomic variation in immune suppressive pathways has revealed that TGFBR2 is associated with prognosis in TNBC patients after chemotherapy [52], which is consistent with the results of pathway analysis in this study.
Limitations: This analysis provides insights about the potential link between genetic susceptibility and the tumor immune microenvironment. However, limitation of the study must be acknowledged. Our study relied on publicly available information on genetic susceptibility variants, some of which may be prone to errors in models used for variant calling. More importantly, our study evaluated all the genes containing genetic variants associated with breast cancer. We took this approach because with the exception of very few studies, GWAS have not focused on breast cancer type-specific. The general design has been a case-control approach.
Our design strategy has followed the same approach by comparing gene expression levels between patients with BLKIA or BLKIS and the controls. It is conceivable that some of the variants and the expression of associated genes may be breast cancer type or subtype-specific, a limitation beyond the scope of this investigation, which we readily acknowledge. This study focused on Caucasian women, specifically because both GWAS and genomics have largely been conducted in women of European ancestry. To the extent that gene expression can be population-specific, genetic variants may confer population-specific risks, and immune responses to pathogens may be population-specific [53] further studies including other ethnic populations are warranted and is the future direction of our research. We did not study the impact of genetic variants on gene expression. However, other published reports have shown that genetic variants regulate gene expression and contribute to gene expression variation in breast cancer [54-55]. Consistent with those findings, our expectation “though speculative” would be that genetic variants could contribute to variability in response to immunotherapy.
Conclusion
This study associates genetic susceptibility with the tumor immune microenvironment in TNBC and establishes putative functional bridges between genetic variants associated with an increased risk of developing breast cancer and the immune modulated pathways they regulate in TNBC. This study sets a foundational framework for future investigations delineating the role of common genetic variants in immunomodulation in this aggressive and often lethal disease in different ethnic populations and funding.
More BJSTR Articles : https://biomedres01.blogspot.com/



No comments:
Post a Comment
Note: Only a member of this blog may post a comment.