Metabolomic Responses of Yersinia enterocolitica Under Acid Stress
Introduction
Foodborne pathogens such as Salmonella Typhimurium, Escherichia coli, Yersinia and other enteric bacteria must overcome several environmental stressors and barriers involved in food production, storage, and host defenses [1]. The enteropathogenic Yersiniae, Y. pseudotuberculosis and Y. enterocolitica, are fecaloral pathogens known to cause a range of invasive gastrointestinal diseases. Furthermore, the bacteria are known for their ability to grow at low temperatures and in acidic environments [2,3]. It has been reported that the pathogenic Yersinia species have also been studied as a model bacterium to understand bacteriahost cell interactions and active pathogen evolution [4-6]. Many foodborne pathogens have developed survival systems that protect them against acidic environments in the stomach, but how these pathogens survive in acidic conditions remains largely unknown [7].
Although transcriptomic and proteomic studies have provided insights into the stress responses of Yersinia, limited metabolomics data exists for some strains of this group [8]. There is a tremendous improvement in food safety and quality research, and application of metabolomics at a holistic overview of the food system, thus improving the overall objectives of ensuring food safety and quality [9]. The high-throughput technique of metabolomics is one of the most recently introduced ‘omics’ that aims to analyze metabolites in a given biological sample [10,11]. Applications of metabolomics include the study of small molecules in a biological system that participates in the metabolic reactions responsible for cell growth, survival, and other normal cellular functions [12- 14]. The two leading analytical approaches to metabolomics are mass spectrometry (MS) and nuclear magnetic resonance (NMR) spectroscopy.
NMR data are highly reproducible and quantitative over a wide dynamic range and are unmatched for determining structures of unknowns [15]. NMR spectroscopy is increasingly renowned for its efficacy, non-invasiveness (non-destructive), throughput and linearity [16]. Moreover, such spectroscopy also provides structural, chemical-kinetics and other information in multidimensional applications [17]. NMR spectroscopy-based metabolomics have been used to study a wide range of biological systems such as tissues [18,19], biofluids [20], mammalian cell cultures [21], plants [22], and bacteria [23,24]. Currently, NMR application has been broadly used and reported for untargeted metabolite profiling of complex food mixtures to address issues of food safety and quality [25,26].
Additionally, a novel NMR-based metabolomics strategy for rapid discrimination and identification of several bacterial species that relies on untargeted metabolicprofiling of supernatants from bacterial culture media [27]. The overall objective of this paper is to investigate the potential of proton nuclear magnetic resonance (1H-NMR)-based metabolomics for the characterization and assessment of Yersinia responses mechanisms to acid stress. Currently, there are limited reports on metabolomic studies examining Yersinia enterocolitica exposure to different environmental stressors including low pH levels. Therefore, in this study, the potential of 1H-NMR metabolomics approach to determine stress responses to acidity in Yersinia was examined to provide in-depth knowledge and application of how foodborne pathogens tolerate the external stresses.
Materials and Methods
Bacterial Strain and Culture Conditions
Yersinia enterocolitica (NR-214) was a gift from Biodefense and Emerging Infections Research Resources Repository (BEI Resources; Manassas, VA). Cultures of Y. enterocolitica were prepared in tryptic soy broth B; Sigma-Aldrich, St. Louis, MO) from frozen stocks and incubated at 30°C overnight and processed as previously described [2]. Briefly, fresh cultures (40ml) were prepared for propagation in TSB at a 1:10 dilution from overnight cultures. Inoculated cultures were grown at 30°C, shaking overnight. After growth, the pH of culture media was adjusted to 3.0 using hydrochloric acid 135 (HCl). After the incubation in acidic media or normal media (control), samples were placed on ice to arrest the metabolism.
Sampling and Metabolite Extraction
The metabolite extraction was performed as previously described [28] with slight modification. Briefly, after 3 h of incubation period, the culture was placed on ice and centrifuged (3,219 xg for 10min at 4°C) for cell harvesting. The cell pellet was washed with an equal volume (40ml) of 1xphosphate buffered saline (PBS) with the same centrifugation parameters (3,219 x g for 10 min at 4°C). The resulting pellet was processed to extract the comprehensive metabolites. Briefly, the washed pellet was resuspended in 1ml of the 100% methanol pre-cooled to -20°C. The suspension was vortexed for 30 seconds and sonicated for 15 seconds. The sonicated suspension was then centrifuged at 17,949 x g for 10min at room temperature to remove cellular debris. The supernatant was collected and dried down using a vacuum centrifuge overnight at 30°C. The dry extract was then stored at -80°C until NMR analysis.
1H-NMR Data Acquisition
Before the analysis, cell extract was reconstituted in 700μl of Deuterium oxide (Sigma- Aldrich, St. Louis, MO) containing 0.025% (w/w) Trimethylsilyl propanoic acid (TSP; Sigma-Aldrich, St. Louis, MO) and centrifuged (17,949xg, 10min, room temperature) to clear any particulate debris. After centrifugation, 650μl of supernatant was transferred to an NMR tube (Sigma-Aldrich, St. Louis, MO). Three replicates were analyzed per experimental group. Onedimensional high-resolution 1H-NMR spectra were acquired on a Bruker Avance II 400MHz spectrometer (Bruker Analytik, Rheinstetten, Germany). All data were collected at a temperature of 300 K using the NOESYPR1D 143 pulse sequence. The experiments were run with 16 dummy scans, 128 acquisition scans and a relaxation delay of 2 seconds. The spectral width was 16 ppm, and 32K data points were collected. All free induction decays were subjected to an exponential line-broadening of 0.3 Hz and zerofilling to 64K points. Upon Fourier transformation, each spectrum was manually phased, automatically baseline corrected, and referenced to the internal standard TSP at 0.0 ppm using TopSpin v3.5 software (Bruker Analytik, Rheinstetten, Germany).
Pre-processing and Data Analysis
The residual H2O 1H-NMR resonance between 4.6 and 5.0 ppm was excluded from analyses along with the extraction solvent peak (methanol, 3.35ppm). Each spectrum was referenced (aligned) and normalized to the TSP reference peak at 0.0ppm. Spectra were then stacked and binned from 0.5 to 10ppm. A table of the average sum integral values for the user defined bins was generated using ACD NMR Processor v12.01 then exported to Microsoft Excel. All spectra integrations were combined into a single excel file then imported into MetaboAnalyst tool suite for multivariate analysis (https:// www.metaboanalyst.ca). Each spectrum was normalized using Probabilistic Quotient Normalization (PQN) to account for sample dilution effects, thus, facilitating comparison analyses of samples [29].
Pathway Analysis
Metabolites were identified based on proton resonances that correspond to chemical shift assignments of known metabolites indexed in the profiling software Chenomx (version 8.3, Alberta, Canada) and the Human Metabolite Database Data Bank (HMDB; http://www.hmdb.ca). Analysis of metabolic cycles was carried out using the freely available online software MetaboAnalyst 4.0, which utilizes the KEGG database for the identification of important metabolic pathways and subsequent biological interpretations. For this data processing step, abundant dysregulated metabolites were filtered according to their VIP score >1.
Results
Metabolic profiling of acid stressed and non-stressed Y. enterocolitica revealed a few changes to the metabolome. Figure 1 shows a comparison of the representative spectral profiles for Y. enterocolitica cultures grown at neutral pH (6.8) versus and cultures adjusted to an acidic environment (pH 3). The identified metabolites are designated as shown correspondingly. Samples were binned and subjected to PLS-DA, which was used 197 to visualize differences between the samples (Figure 2A). According to the PLS-DA scores plot, there was a 91.8% difference (PC-1) in the metabolomic profiles of control and acid-stressed Y. enterocolitica culture extract. We utilized variable importance in projection (VIP), a weighted sum of squares of the PLS loadings considering the amount of explained Y-variation in each dimension, to determine important (VIP score ≥ 1) features/metabolites owing to the model discrimination from the PLS DA plot. To identify specific compounds that contribute to this difference, VIP scores plot (Figure 2B) and heat map (Figure 3) were employed.
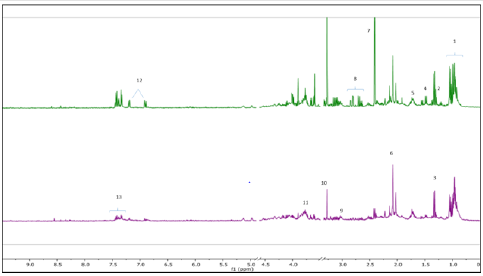
Figure 1:Representative 1H NMR spectra of Y. enterocolitica intracellular metabolic profile. Control (top spectrum), acidstressed (bottom spectrum). Major metabolites identified 1: BCAAs, 2: LDL/VDL, 3: Lactate, 4: Alanine, 5: Leucine, 6: N-acetylglycoproteins, 7: Succinate, 8: Aspartate, 9: pyroglutamate, 10: Betaine, 11: Glutamate, 12: Tyrosine, 13: Phenylalanine.
The potential discriminatory metabolites identified from VIP scores are derived from PLS-DA modeling of complete data matrix and resulted VIP scores for top 23 metabolites shown in increasing order of VIP score values to highlight their discriminatory potential. Branched-chain amino acid (BCAAs) and N-acetyl glycoproteins showed higher levels in the acid-stressed groups. Based on the VIP scores plot betaine, succinate, phenylalanine, and aspartate were mainly decreased for the acid-stressed group. Increased levels of BCAAs are observed for cultures exposed to acidic conditions (Kaisera and Heinrichs 2018). Figure 3 depicted the representative box-and-whisker plots of relative concentrations for selected metabolites from the acid-stressed and non-stressed Yersinia enterocolitica. Principal component analysis (PCA) and partial least squares discriminate analysis (PLS-DA) score plots were used to evaluate the model quality, which is assessed using the goodness of fit parameter (R2) and cross-validation predictive ability (Q2).
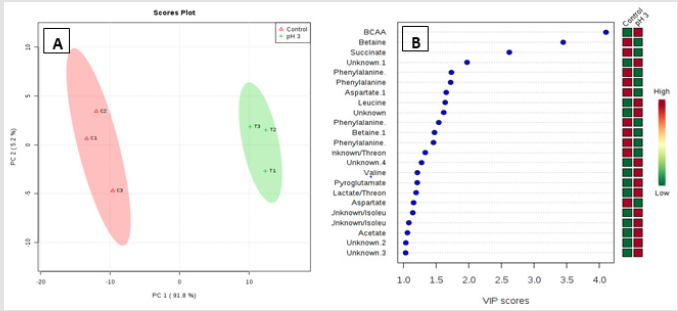
Figure 2:Important features identified by PLS-DA. The colored boxes on the right indicate the relative concentrations of the corresponding metabolite in each group under study. Multivariate analysis consisting of PLS-DA scores plot of all metabolite features (A)and VIP scores plot (B). Projection to latent structures discriminant analysis (PLS-DA) analysis of the estimates of the absolute peak intensities from 1H NMR using all the metabolites/metabolite peaks that is extracted from the spectra. The pH-stressed and non- stressed Yersinia enterocolitica samples clearly separated from each other in PC-1 axial (91.8%). Variable Importance in Projection (VIP) scores are calculated for each components derived from PLS-DA modeling of complete data matrix, and resulted VIP scores (VIP score >1).
Figure 3:Box-and-whisker plots of relative concentrations for significantly changed metabolites in acid-stressed and nonstressed Yersinia enterocolitica. Data were normalized and scaled; and in the y-axes, the concentrations are represented as relative units and the x-axes are the sample group (red = control, green = acid stressed Y. enterocolitica).
Ideal models will have R2 and Q2 values near 1, although in biological systems this is rarely observed due to their variability. In general, a Q2 > 0.5 defined from the metabolomics data is considered good (Figure 4). The color-coded tree line of the dendrogram chart (Figure 5) indicated functional relationships among variables and samples. The data displayed to provide a novel means to understand biological relationships. The clustering shown on Figure 5 as dendrogram chart (distance measure using Euclidean, and clustering algorithm using ward.D) indicated that each sample and the sample groups are distinct from each other. The heat map visualized the metabolomic data whereby the relative abundance of metabolites detected in each sample represented with color intensity. As envisaged on Figure 6, it showed that there is a clear distinction of metabolites pattern between pH stressed and non-stressed Y. enterocolitica. The presence and concentrations of certain group of metabolites are distinctly categorized.
Figure 4: Performance of the PLS-DA classification using different number of components. Model overview of the OPLS-DA model for the provided dataset. It shows the R2X, R2Y, and Q2 coefficients for the groups. The red star indicates the best classifier.
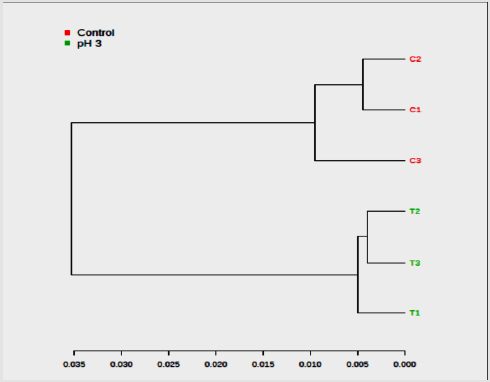
Figure 5:Dendrogram of the hierarchical cluster analysis (HCA) obtained from acid-stressed and non-stressed Yersinia enterocolitica 1H NMR spectra (C = Non-acid stressed and T = acid-stressed Y. enterocolitica).
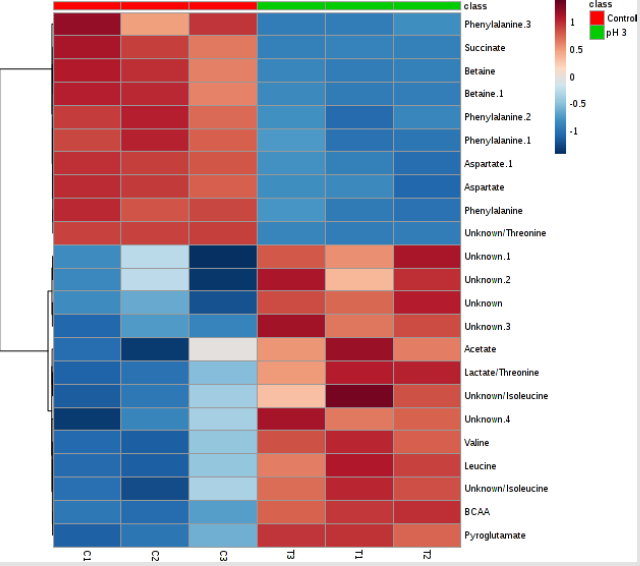
Figure 6:Heat map showing the unsupervised hierarchical clustering of the top 23 differentiating metabolites/features across the control and acid-stressed (pH 3) of Yersinia enterocolitica groups. Each column is labeled with colors according to experimental group.
Each row represents a metabolite feature, and each column represents a sample. The metabolite features levels vary significantly (p < 0.01) across the samples. The compounds that have a VIP score greater than one are enriched in several metabolic pathways (Figure 7). For pathway analyses, the hypergeometric test and relative betweenness centrality algorithms were employed using the MetaboAnalyst 4.0 software. The software provided a fit coefficient (p) from pathway enrichment analysis and an impact factor from pathway topology analysis for each analyzed pathway. The mapping of different metabolic pathways is shown in Figure 7. Exposure to acidic pH indicated changes to protein synthesis, BCAA metabolisms, and other amino acid metabolisms including phenyalanine, alanine, aspartate, and glutamate metabolism. Furthermore, control and acid-stressed cultures could be differentiated by decreased levels of aspartate, phenylalanine, succinate, and betaine (Figure 3).
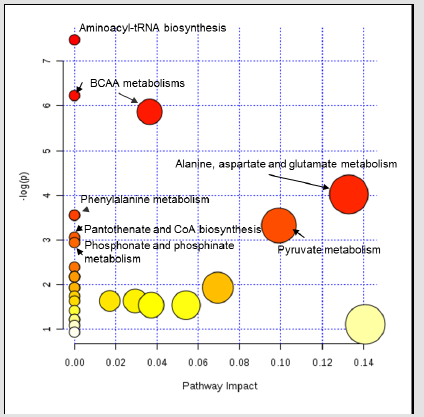
Figure 7:Impact of metabolic pathways for Y. enterocolitica cultures after exposure to acidic conditions (3-pH) as detected by the bioinformatics software, MetaboAnalyst 4.0 (Escherichia coli K-12 MG1655 metabolism from KEGG pathway database was used as a reference). Circle sizes indicate relative pathway impact on the organism’s overall metabolism, while circle color represents relative significance of the pathway to metabolite changes found in the study.
Discussion and Conclusion
Bacteria sense a variety of physical and chemical signals in their environment and use this information to coordinate an adaptive response that promotes their growth and survival. These signals include nutrients such as sugars, lipids, metals, and amino acids, and their depletion triggers upregulation of high-affinity nutrient acquisition systems and/or biosynthesis pathways [30]. Gastrointestinal bacterial pathogens evolved elaborate mechanisms to cope with excursions into acidic environments. Acid survival mechanisms are remarkably different among different pathogens. To cope with different degrees of acidic environmental stress, several acid survival systems have evolved in enteric bacteria [31,32]. The two gastrointestinal pathogens, Y. pseudotuberculosis and Y. enterocolitica, transmit to humans after the ingestion of contaminated water or food.
Like many foodborne pathogens, they have developed different survival mechanisms that protect them against acidic conditions for successful colonization and infection [31,32]. Metabolomics is on the verge of potentially making major impacts including in the areas of foodomics. Previous studies have characterized the metabolic response of different foodborne pathogens in acid environment bacteria [33-35]. It has been suggested that altered metabolites or metabolomics profiles could predict specific metabolic diseases with high accuracy and help to understand related fundamental mechanisms as well as affected metabolic pathways [36]. Untargeted metabolomics measures the levels of thousands of metabolite features in a single analysis, providing a snapshot of metabolism at the systems.
Several visualization techniques have become common in global metabolomic data analysis such as scores and loadings plots, heat maps, scatter plots, volcano plots and recently designed cloud plots level [37-39]. Metabolic analysis reveals evidence for branched chain amino acid catabolism crosstalk and the potential for improved treatment of organic aciduria. Deciphering regulatory circuits that rely on global metabolites enables intuitive understanding and monitoring of cellular decision-making processes [40] results showed that under conditions of low pH, increased branched-chain amino acid biosynthesis protein acetylation were predominantly observed. The paradigm for acetylation in bacteria involves the acetylation of acetyl-CoA synthetase, whose activity must be tightly regulated to maintain energy charge homeostasis [41].
The branched-chain amino acids represent important nutrients in bacterial physiology, with roles that range from supporting protein synthesis to signaling and fine-tuning the adaptation to amino acid starvation [9]. Leucine, isoleucine, and valine are essential amino acids termed branched-chain amino acids (BCAA) due to its aliphatic sidechain. In several pathological and physiological conditions, increased BCAA plasma concentrations have been described [42]. Bacteria synthesize BCAAs through a conserved pathway that is present in fungi and plants, but absent in mammals [43]. Thus, the importance of BCAAs for bacterial physiology stems from their integration with central metabolism, their requirement for protein synthesis, and their requirement for environmental adaptation via Branched-Chain Fatty Acids (BCFAs) synthesis in Gram-positive bacteria [44]. The role of carbohydrate metabolism in acid survival of enteric bacteria remains largely unknown [45].
More is known about the important connections between amino acid metabolism and acid survival in bacteria. Several acid resistance systems, such as glutamate, arginine, or lysine-dependent, have been described in E. coli [46]. In alignment to our results, it has been reported that the enzyme aspartate Ammonia Lyase Or Aspartase (AspA), which is involved in aspartate metabolism by catalyzing the deamination of L-aspartate to form fumarate and ammonia, plays a role in acid survival in Y. pseudotuberculosis [10]. AspA increases acid survivability of bacteria by producing ammonia from aspartate as demonstrated by mutational and in vitro enzyme activity studies. Interestingly, this aspartate dependent acid survival pathway appears to exist in Y. enterocolitica and other foodborne pathogenic bacteria including E. coli O157:H7 and Salmonella enterica, given that the addition of aspartate into culture media alsoincreases their survivability at low pH [10].
The decrease level of aspartic acid and phenylalanine in our results may have been a consequence of both reduced viability and growth after 3 hours of exposure to an acidic environment. We also observed a significant decrease in the osmoprotectant compound betaine from the VIP scores plot. It has been previously indicated that the accumulation of betaine increases the volume of water in E. coli and increases the growth rate of osmotically stressed cells [47]. Likewise, exposure to lethal levels of pH seemed to have resulted in dysregulation of osmotic homeostasis of Yersinia from our study. In some previous report, Glutamate has been indicated to play a central role in a wide range of metabolic processes in bacterial cells. The Glutamate Decarboxylase (GAD) system has been implicated in acid tolerance in several bacterial genera. This system facilitates intracellular pH homoeostasis by consuming protons in a decarboxylation reaction that produces γ‐ aminobutyrate (GABA) from glutamate [48].
Acid exposure induces specific metabolite accumulation in bacterial cells. There are reports of increased valine and isoleucine levels in Streptococcus mutans exposed to acidic conditions [49]. They showed that upregulation of biosynthetic genes such as ilvC and ilvE occurred under acid stress conditions. Additionally, these amino acids can be used as an energy source for bacteria, and they are precursors of branched-chain fatty acids, predominant membrane fatty acids [44]. Moreover, significant changes observed for branched-chain amino acids in this study suggest acid-related stress responses of Y. enterocolitica as key co-regulators of virulence. In addition to branched-chain amino acid metabolism, protein acetylation increased in response to acid stress after 3 hours. Protein acetylation is a widely used protein modification in prokaryotic organisms. Certain acetylases in E. coli and Salmonella have been found to be involved in basic physiological processes, such as primary metabolism, chemotaxis, and stress responses [50].
Protein acetylation impairment in cobB and yfiQ mutants greatly decreased bacterial tolerance to cold, hot, high-salt, and acidic environments for Y. pestis [50]. In alignment to this report, Y. enterocolitica may also upregulate the acetylation of proteins in response to low pH environments. Foodborne pathogens like Yersinia enterocolitica may encounter a wide range of pH environments as the pathogen pass through the oral cavity, gastric transit, and its survival and growth in food matrices. In conclusion, 1H-NMR metabolomics revealed significant information to the metabolomic profiles of Yersinia enterocolitica grown associated with neutral pH versus acidic pH milieu. Differences of metabolite levels observed in this study indicated disrupted protein metabolism, membrane composition, osmotic regulation, and energy metabolism.
This study revealed additional information on acid stress responses of Yersinia, and future work in this area should be directed toward delving deeper into the changes induced in the intracellular and extracellular metabolite pool by similar conditions, as well as investigating the effect of other organic acids commonly encountered in food environments. Results obtained from this study and others including genomics, and transcriptase may highlight new or improved ways to prevent foodborne pathogens from surviving in food products.
For more Articles on : https://biomedres01.blogspot.com/



No comments:
Post a Comment
Note: Only a member of this blog may post a comment.