Epigenetic concerns in therapy for Chronic Obstructive Pulmonary Disease
Introduction
COPD (Chronic Obstructive Pulmonary Disease) is a prevalent chronic adult disease that, by 2020, will be the world’s 3rd leading cause of death. COPD is characterized by Mucociliary dysfunction, lung inflammation, airway fibrosis, and alveolar destruction [1]. There are many factors at hand, such as chronic inflammation, the theory of elastase/anti-elastase, apoptosis, the balance of oxidantantioxidant, and infective repair that cause COPD pathogenesis [2,3]. Cigarette smoke contains over 4700 organic compounds and 1014 free radicals/oxidants, making it the primary source of COPD and inflammation [4]. It stimulates multiple redox-sensitive transcription factors such as NF-kB (nuclear factor kappa-B), most important to increased pro-inflammatory cytokine and chemokine assembly in COPD [5,6]. Post-translational changes in histone and other DNA proteins are known as epigenetic alterations [7]. Without altering the primary gene sequence, DNA methylation and histone methylation can influence gene expression such as post-translational histone protein modifications are acetylation, phosphorylation, and ubiquitination (H2A, H2B, H3, and H4) [8].
Methylation of DNA, which is catalyzed by DNA Methyltransferase (DNMT) family members, which is also the primary epigenetic process in methyl groups are attached to cytosine residues in Cytosine Phosphate-Guanine (CPG) [9,10]. Hypermethylation of DNA in CpG island gene promoter regions result in gene silencing, while hypomethylation results in DNA transcription activation [9]. Histones are essential protein components of chromatin. Chromatin consists of H-linker and octamer protein containing two copies of each H2A, H2B, H3, and H4 core histone where DNA is wrapped [11]. H3K4me3, H3K9me3, and H3K27me3 are modifications of histones that play a significant role in controlling genes. The activation of gene expression with H3K4me3, but gene expression is associated with H3K27me3 and H3K9me3 [11].
DNA Modification in COPD
The same DNA sequence can be found in all of an organism’s cells. Thus, with the aid of epigenetic control, the genome is organized into ‘heterochromatin’ (compact) and ‘euchromatin’ (accessible) regions to ensure the right transcriptional program in a particular cell type. Changes in DNA alteration may explain some of the considerable variations in smokers’ vulnerability to developing COPD. Indeed, 349 Cytosine phosphate Guanine (CpG) sites were confirmed to be significantly associated with the intensity of COPD and the reduction in lung function by large-scale genome-wide investigations of DNA methylation marks (27,000 genes) [12]. Cigarette smoking (CS) condensate can cause multiple CpG targets in small human airways and bronchial epithelial cells to hypo or hypermethylation [13]. Changes in DNA methylation patterns have been found to enhance histopathology based on the experiment carried out on mice for smoke exposure. Similarly, DNA methylation caused by various dangerous elements may increase the sensitivity of COPD to a subsequent insult. This is confirmed by the transparency of pre-birth cigarette smoke affects explicit CpG DNA methylation in detoxification characteristics worldwide and performance. This also leads to constant effects, such as decreased lung work in young people [14,15]. Previously Georgiou, et al. [16], proved that chronic smoking is associated with increased methylation of a known tumor suppressor, p16 promoter, or aryl hydrocarbon receptor repressor; changes in DNA methylation status following smoke exposure can explain the altered expression in DNMT (DNA methyltransferase).
This subject has only been marginally debated about lung cancer, but not concerning COPD. CS exposed human bronchial epithelial cells had lower levels of DNMT1 and higher levels of DNMT3b [13]. On the other hand, CS elevated DNMT1 levels in A549 cells, while DNMT3b downregulation triggered synuclein-gamma demethylation, a prometastatic oncogene. DNMT1, DNMT3a, and DNMT3b proteins are significantly elevated in patients with lung tumors [17]. In addition, nitrosamine ketone (NNK) obtained from tobacco-specific carcinogenic nicotine-induced nucleus aggregation of DNMT1, accompanied by hypermethylation of the tumor suppressor gene in mice and lung cancer patients who regularly smoked [13]. Polycomb group (PcG) proteins were discovered for the first time in Drosophila melanogaster, and they represent a novel epigenetic control region. The Polycomb target gene RUNX3, a tumor suppressor gene, is associated with promoter methylation and CS [18]. Besides, it has been demonstrated that CS condensate silences tumor suppressor miRNA, miR-487b expression epigenetically. In lung epithelial and lung cancer cells following CS treatment, PRC-mediated DNA methylation and altered nucleosome location within the miR-487b region were observed [19].
DNA Methylation and COPD
DNA methylation is revealed to play a critical function in the existence and output of COPD; CS modifies the DNA methylation by triggering inflammatory responses and causing diseases such as COPD [12]. Macrophages are innate immune cells that directly affect the polarization of innate and adaptive immunity and eliminate by lung macrophages of bacteria [20]. Usually, COPD arises with upper lobe predominance, and tests have demonstrated anatomical variation in ventilation and oxygenation of the upper and lower lung lobes [21]. The main ways of modifying epigenetics DNA methylation shows in (Figures 1 & 2).
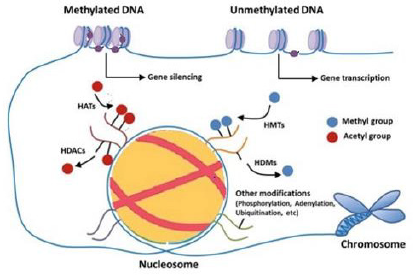
Figure 1: The main ways of modifying epigenetics. DNA methylation, histone methylation are involved in most epigenetic modifications. The action of DNA methyltransferases can regulate DNA methylation (DNMT). Modifications of histones are mainly expressed in acetylation. Mediated by histone methyltransferases (HMTs) and histone demethylase (HDMs), and histone deacetylases (HDACs).
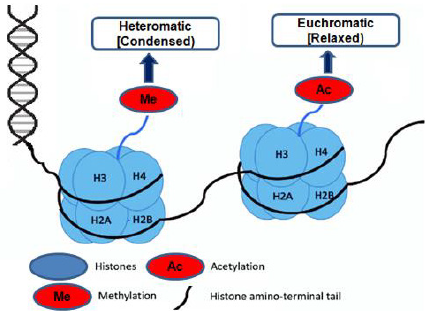
Figure 2: Schematic drawing about chromatin remodeling of histone methylation and acetylating.
Targeting DNA Alteration for COPD Therapy
In COPD, DNA changes are observed by replacing cytidine within the DNA strain. Nucleoside analogs such as5-aza-2-deoxycytidine and 5-azacytidine act as DNMT inhibitors. Most of the substrate analogs are, modified by DNMTs analogs through covalent binding, which then inhibits the functioning of enzymes, resulting in hypomethylation of DNA. The PRC2 methylation binding sites are altered or reversed using azacitidine, resulting in (5-azacytidine) non-metastatic status [22].
Histone Modifications
Histone modifications were grouped into eight categories based on the type of amino acid and modified into methylation, methylation of arginine, acetylation of lysine, serine, ADP ribosylation, threonine phosphorylation, proline isomerization, arginine deamination, sumoylation, and ubiquitination [23,24]. The induction of a double-strand break at the DNA damage foci, histone acetylation, and methylation plays an essential role in the DNA damage response. Histone H3K9 and H3K56 acetylation and H3K4 and H3K36 trimethylation have been related to active gene transcription (euchromatin). In contrast, histone H3K9 and H3K27 tri-methylation have been linked to silence gene loci (heterochromatin) due to the closure of active chromatin in the gene [25-28].
Role of Histone Methylation in COPD
The modulation of histones is much more significant than DNA methylation. A recent mass spectrometry technique has identified possible novel histone marks in smoke-exposed mouse lungs, including acetylation, mono-methylation, and di-methylation of specific lysine and arginine residues of histones H3 and H4 [29]. Histone H3K27me2 and H3K27me1 were only found in the CS sensitivity community. These findings suggest that Polycomb plays a role in specific gene repression, particularly after smoking [29].
COPD Treatment Based on Histone Modifications
In patients with COPD, effective anti-inflammatory tested by treating with inhaled or systemic corticosteroids. However, even massive doses of corticosteroids showed an immune response in COPD patients. Impaired Histone Deacetylases (HDAC) function has recently been analyzed in-depth as one of the pathways contributing to corticosteroid tolerance. The activity of HATs was inhibited by activating Glucocorticoid Receptors (GR) which binds to the coactivators (Histone Acetyltransferase) [30,31]. Theophylline has been used for decades to treat airway conditions and was recently discovered as an activator of HDAC. In COPD patients alveolar macrophages, theophylline restores HDAC activity and steroid response [32]. In CS-exposed mice, a low dose of theophylline increased the anti-inflammatory effects of dexamethasone while also restoring HDAC2 movement [32]. The pharmacological activation by a small molecule activator, sulforaphane, of the antioxidant NRF2 can be a valuable technique for preventing and treating COPD and is currently being tested in patients with COPD in phase II clinical trials [33].
By deacetylating lysine residues that would or else be targeted for ubiquitin-mediated proteasomal degradation, functional HDAC2 is likely to inhibit NRF2 degradation [34]. In peripheral lung tissues and alveolar macrophages, HDAC2 activation was shown to be lower in patients with COPD when the enzyme was S-nitrosylated. Treatment with sulforaphane nitrosylated HDAC2 resulted in simultaneous activation of NRF2 and augmented intracellular glutathione levels in alveolar macrophages [35].
Conclusion
Dysregulation of DNA methylation in COPD-related genes, HDAC2 function, and miRNAs have all been shown to cause COPD disease in various studies. Inflammatory reactions are triggered by tobacco smoke and are a central factor in COPD development. It changes epigenetic operations such as gene expression by modifying DNA methylation, altering HDAC2 function by histone modifications, and dysregulation of COPD linked to mRNAs. Our review emphasizes the recent studies of specific techniques, such as Sirtuins, to control the activity of HDAC2 and have a therapeutic effect on COPD. The miRNAs have been shown to decrease COPD in more recent research. Finally, this review can address or help study a greater variety of issues in both diagnosis and treatment methods for COPD.
For more Articles on : https://biomedres01.blogspot.com/



No comments:
Post a Comment
Note: Only a member of this blog may post a comment.