The Complete Chloroplast Genome Sequence and Phylogenetic Analysis of Mulberry (Morus alba Lin.)
Introduction
The chloroplast (cp) is the photosynthetic organelle representing one of the most important organelles in green plants and algae [1]. The origin of chloroplasts can be dated back to about 390,000 years ago [2]. The mainstream view is the theory of endosymbiosis, which believes that plant chloroplasts originated from ancient cyanobacteria that were symbiotic in primitive eukaryotic cells [3]. In angiosperms, the chloroplast genome (cpDNA) is typically composed of a pair of inverted repeat regions (IRa and IRb), which are separated by a small single-copy (SSC) region and a large single-copy (LSC) region [4]. The contraction and expansion of IR region [5,6] determine the length of the chloroplast genome.
Most cp genomes are 120–220kb in length and contain 120–140 coding genes (Marcelo, Leila, Fraga, & Guerra, 2015; Méndez-Leyva et al). The size of chloroplasts are similar in related specie [7]. The GC content in the chloroplast genome is generally low, about 37% on average, with AT tendency [8]. The chloroplast sequence is wildly used to phylogenetics, species identification, population genetics, and genetic engineering [9-11]. Recently, reports on phylogenetic analysis in rice [12], analysis clarify the taxonomic status of Capsicum L (Elmosallamy et al. 2019) and Korean ginseng [13]. The cpDNA sequence of Moraceae is incomplete and limited. The results of phylogenetic study on mulberry based on nuclear genome redefined the mulberry as eight species [14]. But the conclusions are not enough to reveal the complex systemic origin, evolutionary relationship, and phylogenetic studies of mulberry. DNA molecular markers like indels, SSRs and small inversions [15], ITS regions [16,17] have been used to study genetic and genome diversity and phylogenetic analysis. In this study, In this study, the cpDNA sequences of M. atropurpurea and M. multicaulis were investigated, and a comparative analysis was performed between cultivated Morus and M.mongolica. The genome structure, gene order, repeat sequences, and phylogenetics were analyzed.
Materials and Methods
Plant material, DNA Extraction, and Sequencing
M. atropurpurea (Lunjiao40) and M. multicaulis (Husang32) fresh leaf were collected from National Mulberry Genebank Zhenjiang City, Jiangsu Province. The plants DNA were extracted using CTAB method for complete and high purity DNA to build DNA library following manufacture’s protocol. The DNA concentration was measured by Nanodrop instrument then qualified samples were sent to the Illumina NovaSeq for sequencing.
Quality Control of Sequencing Data
To improve the accuracy of the analysis, the Raw Reads were filtered again according to the following criteria:
1. Removal of the sequenced connectors and primer sequences in reads,
2. Reads with an average mass value less than Q5 were filtered out,
3. Reads with N number greater than 5 were removed. The quality reads after the above checks, called clean reads were subjected to subsequent analysis.
Assembly and Annotation
The SPAdes (3.13.0) software was used for the genome splicing [18], The candidate sequence assembly was determined, annotate, and then GeSeq was used to draw the circular gene map.
Analysis of Repeated Sequences
Long repeats include three types: forward (P), palindrome (P) and tandem (T) repeats, which may promote chloroplast genome rearrangement and increase population genetic diversity. We use vmatch (http://www.vmatch.de/) The software (parameter: minimal repeat size 30bp) finds the scattered long repeat fragments in the chloroplast genome.
Comparative Analysis of Chloroplast Genomes of Morus Species with Other Species
The mVISTA online software in shuffle-LAGAN mode was applied to compare the complete chloroplast genomes of cultivated Morus species with four representatives. M. multicaulis (husang32) was used as a reference. Also, different families Arabidopsis thaliana (NC000923.1), Fragaria chiloensis (JN884816), Oryza sativa (NC- 008155), Nicotiana tabacum (Z00044) were analysed together. The software (parameter: minimal repeat size 30bp) finds the scattered long repeat fragments in the chloroplast genome.
Phylogenetic Analysis
The MEGA X software was used to determine the phylogenetic relationships between Morus species by the maximum likelihood (ML) and neighbor-joining (NJ) methods.Data on the cpDNA of Morus species are available, including those from including those from NCBI M. indica (NC-008359), M. mongolica (KM491711), M. notabilis (KP939360).
Results
Analysis of Sequencing Data and Quality Control
An overview of the chloroplast sequencing reads derived from the M. multicaulis M. atropurpurea libraries is listed in Table1. A total of 30417184 and 32296130 raw reads were obtained from M. multicaulis and M. atropurpurea, respectively. After quality control check on the raw reads, 30374110 and 32246870 clean reads were obtained from M. multicaulis, and M. atropurpurea respectively. In addition, the Median Phred Quality Score as well as the Read Cycle in mulberry plants are shown in Figure 1.
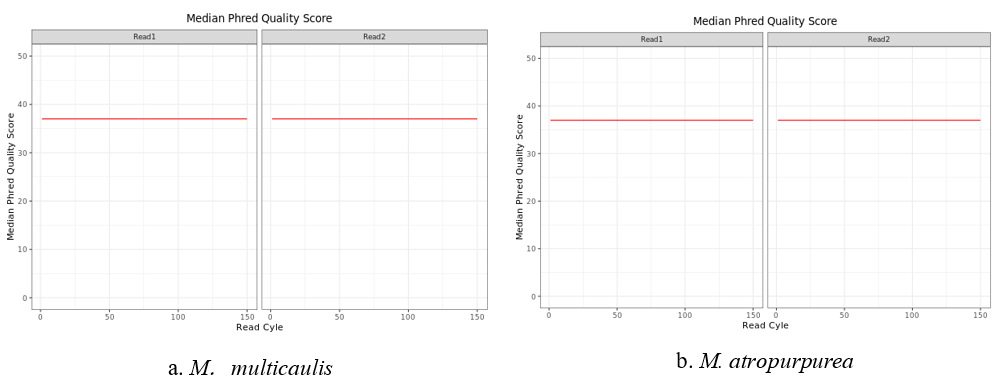
Figure 1: Rawdata of M. multicaulis and M. atropurpurea.
Genome Structural and Content
The cpDNA sequence was determined (Figures 2 & 3) In our studies, the length cpDNA of M. multicaulis is 158,817 bp. longer than M. atropurpurea 158,776 bp (Table 1). M. multicaulis with, a circular double-stranded DNA composed of two identical IR regions (25,551). bp), an LSC region (87,880 bp) and an SSC region (19,835 bp). M. atropurpurea (Lunjiao40) also has a typical quadripartite structure with 158,776 bp long, IRa and IRb with 25,667 bp in length separated by LSC region (87,670 bp) and SSC region (19,750 bp). The GC content of M. multicaulis chloroplast genome is 36.24%., LSC (33.90%) and SSC (29.24%) regions which are lower relatively lower, compared to the IR region (42.99%) (Table 2). The GC content of the chloroplast genome is very close, and no changes were found to occur in the IR region of the seven mulberry species selected. The cpDNA contains 113 functional genes, including 79 protein-coding genes, 30 tRNA genes and 4 rRNA genes. Pseudogenes and ORFs not included (Table 3). The cpDNA can be divided into three categories depends on the function. The first is related to self-replication containing 70 genes, the second is related to Photosynthesis containing 38 genes, and the third is related to fatty acids and amino acids biosynthesis. There was a total of 5 genes with unknown functions. The results is similar to that found in green plants [6].
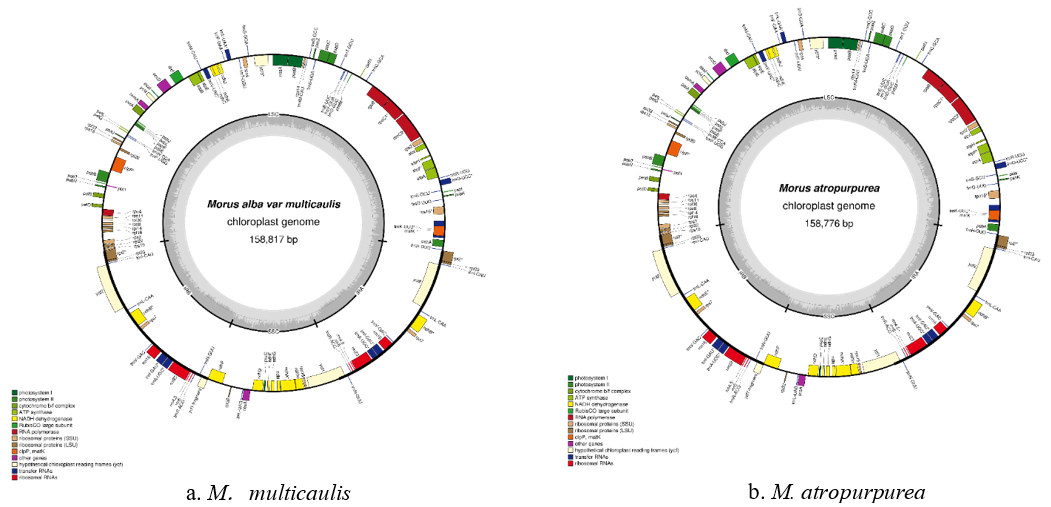
Figure 2: Gene map of the chloroplast genome of M. Multicaulis and M. atropurpurea.
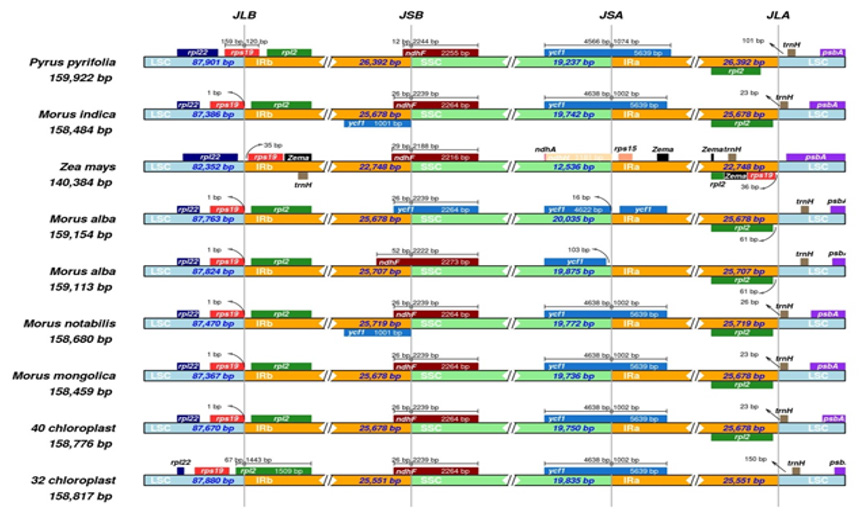
Figure 3: Comparison of the junction between IR and SC regions.32: M.multicaulis.40: M.atropurpurea.

Table 1: Rawdata of Morus.
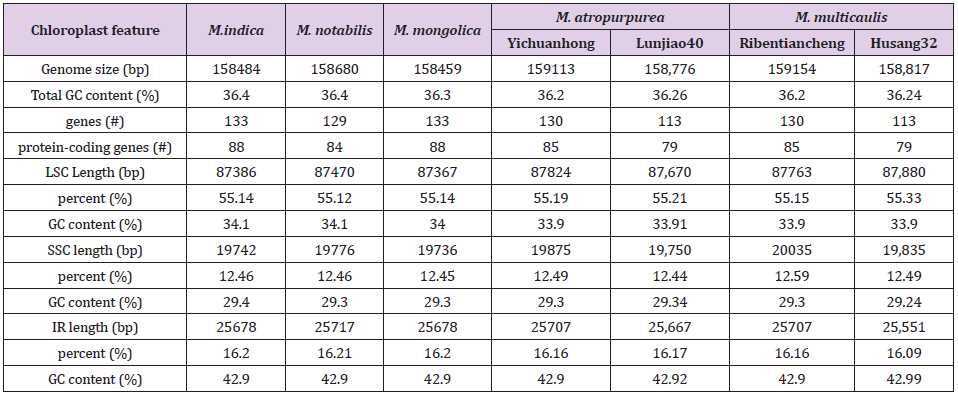
Table 2: Comparison of chloroplast genomes between seven Morus L species.
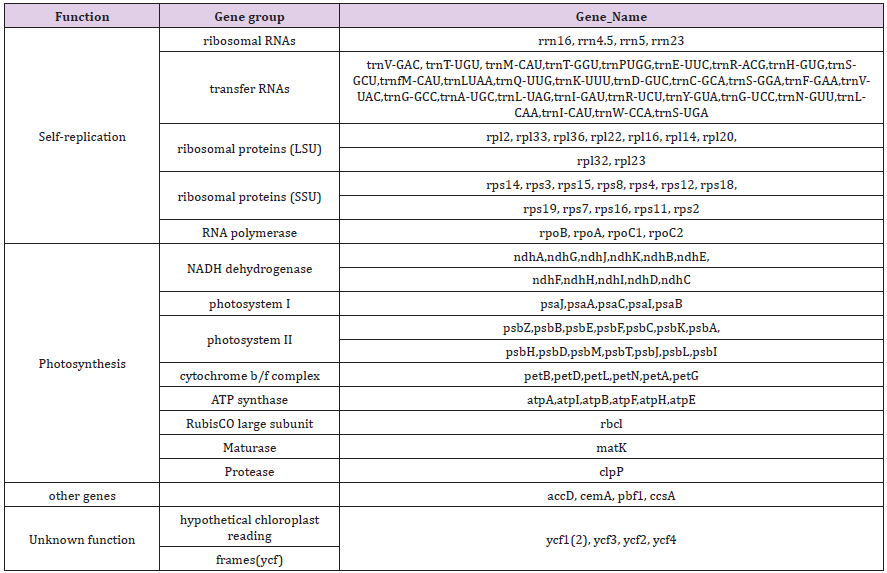
Table 3: Genos present in the chloroplast genome of M. atropurpurea and M. multicaulis.
Codon Usage
All 79 protein-coding genes in the cpDNA of M. multicaulis and M. atropurpurea were encoded by 60,765 codons (Table 4). Codon usages have an AT tendency. For M. multicaulis 63.76% of codons ending with A or T. Amino acid Leu has the most codons usage of 6,435 (10.59%) codons, followed by Ile with 5310. Ser with 4,464 and Glu with 4,464. These four amino acid codons represented one-third of the total. The least Cys had 699 codons (1.15%). AUU is the most frequent start codon (872), and UGA is the most common stop codon (11).
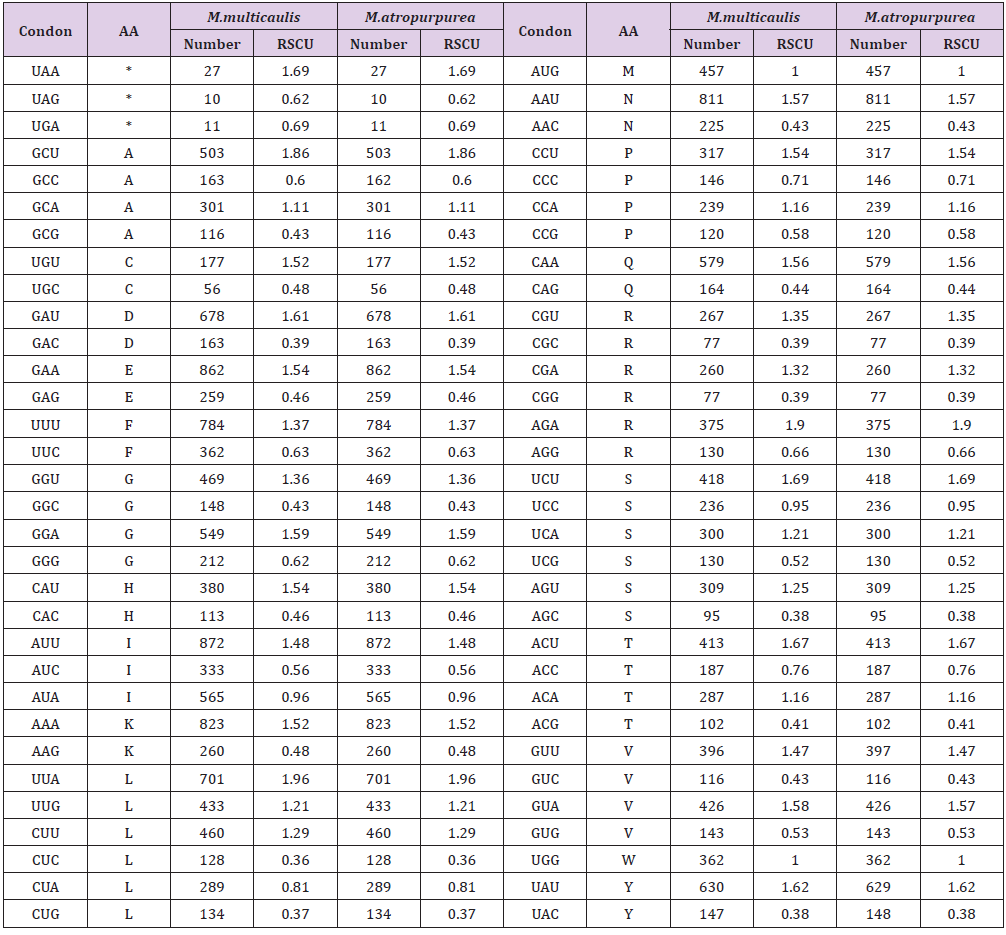
Table 4: Codon usage for M. multicaulis and M. atropurpurea.
Repeat Sequences Analysis
Long repeats include three types: forward (P), palindromic (P) and tandem (T) repeats, which may promote chloroplast genome rearrangement and increase population genetic diversity. The M. atropurpurea with the most extended repetitive sequence located in 133,098 with 25,678 bp, which is 127 bp longer than that of M. multicaulis and the shortest is 31 bp, which is 8 bp shorter than that of M. multicaulis (Tables 5 & 6). This is a phenomenon of genome rearrangement and may be related to genetic diversity.
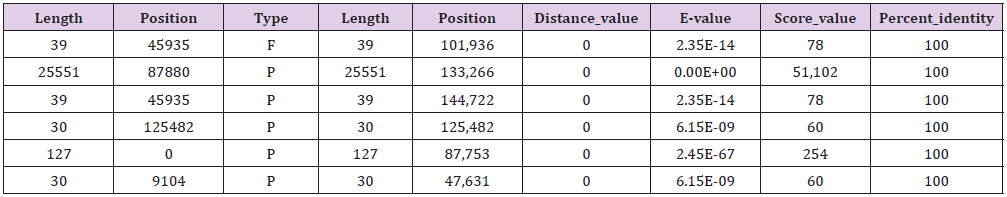
Table 5: M. multicaulis Long repeat sequence analysis.
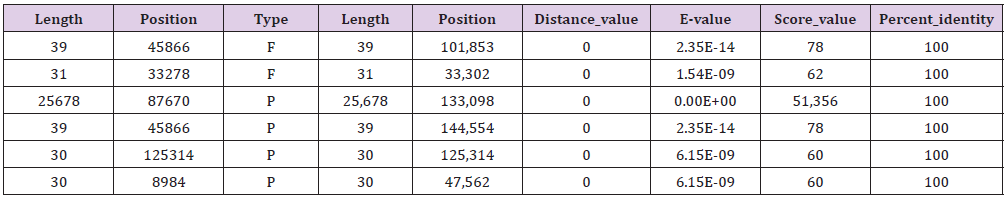
Table 6: M.atropurpurea Long repeat sequence analysis.
Comparison with Other Species Chloroplast Genomes
We compared with others 6 species and two exogenous Pyrus pyrifolia and Zea mays based on the complete cp genomic sequences (Figure 3). The results showed that the IR boundary of Morus chloroplast genome is slightly different. The rp12 gene at the junction of LSC/IRb 67bp in LSC and the rest are in IRb and trnH gene which is 150bp away from the IRa/LSC boundary of M. multicaulis. Pyrus pyrifolia is 101 bp away from the IRa/LSC boundary. The junction IRb/SSC 52bp of ndhF gene is located at IRb, and the rest is located at SSC of M. alba. Ycfl is located in IRa/SSC boundary, resulting in the formation of ycfl pseudogene. The rpsl9 gene is located at the LSC/IR boundary, which resulted in the formation of a rpsl9 pseudogene. These results are similar to previous study findings [19]. We analyzed the chloroplast genome sequences of 7 mulberry species and Arabidopsis thaliana, Fragaria chiloensis, Oryza sativa, Nicotiana tabacum using mVISTA software (Figure 4). Results showed that the chloroplast genome within the genus Morus was relatively similar. The regions with higher variation were distributed in the LSC and SSC regions, and the IR region was more conservative.
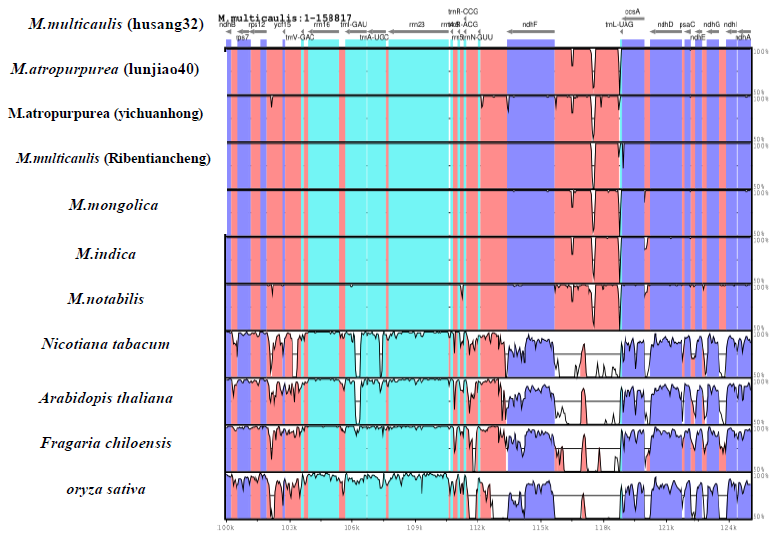
Figure 4: Percent identity plot for comparison of seven Morus L. chloroplast genomes using m VISTA.
Phylogenetic Analysis
Using MEGA-X(10.2.2)software through the ML (Figure 5) and NJ methods (Figure 6) cluster analysis based on the complete chloroplast genome sequences. M. multicaulis (Husang32) and M. atropurpurea (Lunjiao40) are grouped together it’s means they are closely related and diverged from the M. indica and M. notabilis earlier.
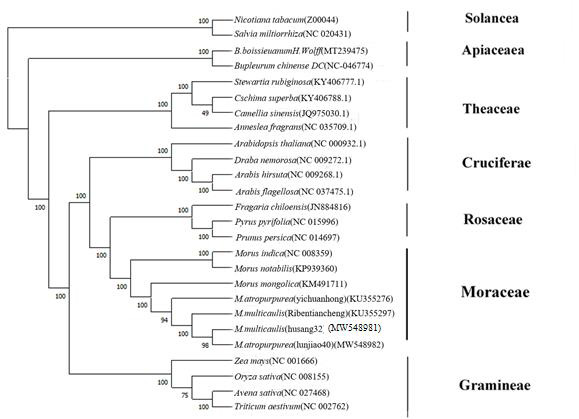
Figure 5: Phylogenetic analysis of the Morus L. using complete chloroplast genome by the ML method.
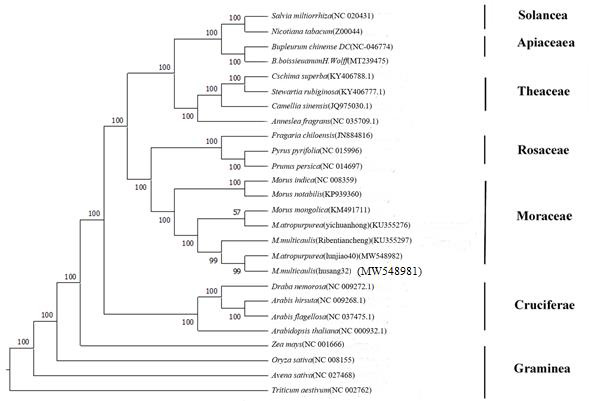
Figure 6: Phylogenetic analysis of Morus species using the complete chloroplast genome by the NJ method.
Discussion
In this study, we collected two cultivated species of Morus L. (M.atropurpurea and M. multicaulis) assembled and annotated the cp genome and performed extensive analyses based on the complete cp genome sequences and amino acid sequences of the annotated genes. The Morus L cp genome is circular contain LSC, IR, SSC. compared with wild species of M. mongolica, M. notabilis and M. indica in the genome sequence, providing more detailed information for phylogenetic studies. The results show that the chloroplast genome length of 7 mulberry species ranges from 158,459 to 159,154 bp. M. atropurpurea and M. multicaulis with two species different accession also have slight differences. "Husang32" and "Ribentiancheng" of M. multicaulis, with "Husang32" 377bp being shorter than the "Ribentiancheng" (Table 1). This result agrees with the gene sequence used by Li Qiaoli [20] on M. multicaulis. Also, M. atropurpurea "Lunjiao40" is 337bp shorter than "Yichuanhong" (Table 2). There were few differences in the length of the IR and SSC regions of the cpDNA from seven species, but most of the differences mainly focus on the LSC region. The results indicated that these species are closely related and confirmed by the phylogenetic tree analysis.
IR expansion/contraction studies reveal the considerable difference even in the same family [13]. It is believed that the borders of the IR region (IRa and IRb) with the LSC and SSC regions, some expansion and contraction may play an important role in evolution [21]. Therefore, it is of great interest to compare IR/SC junctions in different varieties. Based on this, we use 6 mulberry species along with other members, complete chloroplast genome sequences. The results showed that rp12 gene located in the junction of LSC/IRb 67bp in LSC and the rest are in IRb and trnH gene is 150 bp away from the IRa/LSC boundary of M. multicaulis. With ndhF located in the junction of IRb/SSC 52bp at IRb, the rest is located at SSC of M. alba. Based on IR boundary, it could be seen that M. atropurpurea and M. multicaulis are closely related and together also closely related to M. mongolica and M. notabilis. This result agrees with the finding by Li Qiaoli [20].
Usually, cpDNA also tends to AT [7,22], The AT content of the four regions of the seven species (Table 2) shown The IR region has the lowest with an average of only 57.08%, LSC region and the SSC region are 66.02% and 70.67%, M. atropurpurea, and M. multicaulis were slightly higher than other groups. The average of 4 species was about 63.78%, SC regions with a high AT content harbour more variation SSR polymorphisms between M. multicaulis and M. atropurpurea are all involved A or T mutations. The lowest AT content in the IR region maybe because it contains all the rRNA, which is very conservative, and the GC content is high and relatively stable. The rpl21 gene is known to only exist in the plastomes of ferns and bryophytes [23]. This result shows that Morus contains two pseudogenes ycfl and rpsl9. SSR as an important molecular marker used for studying population genetics [24,25]. Which may suggest their usefulness in future evolutionary studies on Morus species. With the Phylogenetic analysis, seven species of Morus gathered together. Both M. atropurpurea and M. multicaulis gather together, which is the closest. This result is similar to Li Qiaoli [20]. In the present research, cpDNA sequences of Morus species are minimal. Our experimental results do not represent the final classification of the Morus system. Therefore, the evolutionary relationship of Morus needs further research to draw more accurate conclusions. Reported here enhance genome information on Morus and contribute to the study of germplasm diversity. These data represent a valuable Phylogenetic analysis source for future studies on Morus populations diversity. Genetic diversity within the chloroplast is considered effective tool for understanding population genetic structure and species evolution. We assembly the full chloroplast DNA sequences of two cultivated species of M. atropurpurea and M. multicaulis and compared with other six wild Morus species. These sequences enabled us to identify evolution divergence time of Morus and can be used for further research of genetic diversity, genetic structure, and genome evolution history of Morus [26,27].



No comments:
Post a Comment
Note: Only a member of this blog may post a comment.