Kinetics Study The Catalyze Mode Of The Formamide Degradation Reaction By Aux Cluster (X=0, 1, 2, 3): Point, Line Or Surface?
Abstract
The related energy and multi-channel CHONH2 degradation reaction potential energy surface under the catalysis of Aux
(x=0, 1, 2, 3) cluster was studied, while its dynamic characterization
has been investigated with density functional calculations. The
geometries were fully optimized with the CCSD(T)//B3PW91 level. We can
conclude two results: [1] The calculated results shown that the main
pathway of the CHONH2 degradation reaction under the catalysis of Aux (x=0, 1, 2, 3) cluster can give the main product P1 (CO+NH3), while the minor product is P1(H2+HNCO) and P3 (H2O+HNC).
We calculated the rate constant of the main reaction pathway, the
calculated dynamic characterization indicating that the rate constants
have the positive temperature dependence. According to the dynamic
results and the energetically intermediates and transition states
involved in the dominant paths, the reaction is expected to be occurred
the most rapidly under the catalysis of Au2. [2] From the
PESs, we can see the present invention, the singlet atom is the best
catalysis and can catalyze the reaction better. The present invention
studies may provide useful information on the issues of the reaction
mechanism and product distributions.
Abbrevations: NBO: Natural Bond Orbital;
ED: Electron Density; DFT: Density Functional Theory; PES: Potential
Energy Surface; RECP: Relativistic Effective Core Potential; IRC:
Intrinsic Reaction Coordinate; SCT: Small Curvature Tunnel Effect
Correction
Introduction
Gold has been enjoyed as the most inert metal for a very long time,
until the pioneer of Haruta [1,2] found that the gold nanoparticles
supported on some metal oxides showed surprisingly high catalytic
activity for small molecule reaction at low temperature. , the Nano gold
catalysis has picturesque growing in the field of heterogeneous
catalysis [3-5]. Technologically, numerous potential industrialization
applications of Nano gold catalysts were developed, such as synthesis of
fine chemicals [6,7], selective hydrogenations [8,9], water gas shift
reaction [10], carbon–carbon bond forming reaction [11], oxidation of
organic compounds with molecular O2 [12,13], pollution and
emission control [14] and fuel cell applications [15,16] Scientifically,
a huge number of theoretical and experimental studies have been devoted
to understand the wide gap between the chemical inertness of bulk gold
and high catalytic activity of Nano gold[17-21]. The photolysis of
formamide vapor has only been reported [22,23]. They studied the
degradation reaction of formamide and gave the three major primary
processes:
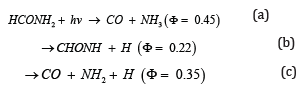
According to their experimental conditions and results of the quantum
yields ɸ, the threshold energy of channel a seems to be the lowest.
Although the pyrolysis of formamide vapor apparently has not studied
previously, several competing channels could also be expected in the
thermal system. There are several reports about the competitive
reactions in thermal systems [24,25], they conclude that the rate
constants increased as the temperature arises. There are still unknown
parameters in the descriptions and the rate constants, these two
problems have not been entirely settled. Especially for complex
bond-breaking reactions, the direct experimental determination of the
threshold energy is almost impossible. Ab initio molecular orbital
methods have developed rapidly and have provided heats of reaction,
potential barriers, molecular geometries, and the vibrational
frequencies of several
intermediates and transition states. This valuable information
can shed light on reaction mechanisms, and ab initio calculations
have been applied to several unimolecular reactions. For complex
bond-breaking reactions, information about the transition states is
required to test the experimental results.
In the past two decades, the power of computational hardware,
software gradually increased and therefore theoretical calculations
became more and more popular and important to investigate
structures, stabilities and activities of catalysts. calculation relies
only on the electron density, which significantly decreases the
computational complexities. It becomes an increasingly powerful
and useful tool to evaluate various systems, predict catalytic
activities, and provide enough accuracy to be compared to
experimental data [26]. So far, DFT calculation was widely used to
investigate the mechanism of gold catalysis and provide fundamental
physical insights into gold catalysis [17,27-28]. For example, selfconsistent
density functional calculations showing that an isolated
Au10 cluster should be able to catalyze the CO oxidation reaction
even below room temperature and to use calculations can analyze
the origin of this effect and suggest that the extraordinary reactivity
can be traced back to special reaction geometries available at
small particles in combination with an enhanced ability of low
coordinated gold atoms to interact with molecules from the
surroundings [17,18]. Based on the molecular orbital analysis and
the DFT calculations, we have discussed the redistribution of in
various bonding and antibonding orbitals and the energy of hyper
conjugative interaction stabilization energy (E(2)) which have
been calculated by natural bond orbital (NBO)[29] analysis using
DFT method to give clear evidence of stabilization originating from
the hyperconjugation of various intramolecular interactions. The
calculations are valuable for providing a reliable insight into the
molecular properties.
In this work, we will give a brief review of theoretical
investigations on CHONH2 degradation reaction catalyzed by
Aux (x=0, 1, 2, 3) cluster, since most of the reviews focused on
experimental results and the reviews of theoretical work were
relatively rare. No major products are given and there is no available
information on product channels, product distributions, and the
reaction mechanism in the experiment. Considering the potential
importance and the rather limited information, we carry out a
detailed theoretical study on the potential energy surface (PES) of
the CHONH2 degradation reaction to
a) Provide the elaborated addition channels on the CHONH2
degradation reaction PESs;
b) Give a deep insight into the mechanism of CHONH2
degradation reactions;
c) Calculate the rate constant of the H-abstraction. We hope
our work will provide some valuable fundamental insights into
title reaction.
Computational Details
All Au clusters and CHONH2 molecule calculations are carried
out using the Gaussian 09 program packages [30]. The geometries
of all the stationaries are optimized using the hybrid density functional
B3PW91 method [31]. As is known, full electron calculations
for Au atom consume too much time, so it is necessary to introduce
the relativistic effective core potential (RECP) to describe the inner
core electrons. The 5s25p65d106s1 outermost valence electrons of
the Au atom are described through the Lanl2dz base set [32, 33].
In order to obtain more reliable energetic data, higher level single-
point energy calculations are performed at the multi-coefficient
correlation method based on quadratic configuration interaction
with single and double excitations (CCSD(T)) [34-38] by using
the B3PW91 optimized geometries. To confirm that the transition
states connect designated intermediates or products, intrinsic reaction
coordinate (IRC) calculation is carried out at the B3PW91 level.
Moreover, the CCSD(T) single-point energies with ZPE corrections
are used in the following discussions.
NBO method [39] is performed to study orbital interactions
supported by Gaussian 09 program. The localized orbital
interactions, involved in non-covalent interactions, are quantified
using second order perturbation theory, in which the secondorder
energy (E(2)) was used to measure the interaction strengths.
We use the Arrhenius formula to calculate the rate constant. The
rate constant of the rate-controlling step along the main reaction
channel was calculated among the temperature range 200~2000
K, which considers the small curvature tunnel effect correction
(SCT) [40,41].
Results and Discussions
For our convenient discussion, the energy of reactants R is
set to be zero for reference. By means of the transition states and
their connected isomers or products, a schematic potential energy
surface (PES) of the most relevant reaction pathways is described
for the CHONH2 degradation reaction catalyzed by Aux (x=0, 1, 2,
3) cluster and the optimized structures of the important stationary
points are plotted in (Figure 1).
The Related Energy
Data on formation enthalpies constitute an excellent means
to establish whether theoretically predicted phases are likely to
be stable, and such data may serve as a guide to evaluate possible
reaction routes. For the exploration of the thermodynamic
feasibility of accessing these compounds from the elements (eq.
(1)) we have also computed the total energies for CHONH2 + Aux
(x=0, 1, 2, 3) in their ground state structures with full geometry
optimization. The adsorption enthalpies for CHONH2 + Aux (x=0, 1,
2, 3) were calculated from the difference in the total energy which
are summarized in Table 1. The results establish unambiguously
that (eq. (1)) expresses endothermic process for CHONH2 + A ux
(x=0, 1, 2, 3) reaction.
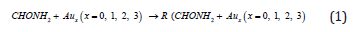
The formation energy for the prototypical CHONH2 +Au1 are
-71.4 and -63.5 kcal/mol respectively, indicating that CHONH2+Au1
is a thermodynamically stable phase at ambient conditions. This has
already been established by a series of experimental and theoretical
studies. Our estimated large positive values for the enthalpy of
formation for the CHONH2+Au1 series also suggest that it might be
possible to degradation compounds by singlet Au catalyst.
Reaction Potential Energy Surface
A schematic potential energy surface of the CHONH2 degradation
reaction obtained at the CCSD(T)//B3PW91 + ZPE level is plotted
in Figure 1a. On the PES for the CHONH2 degradation reaction,
1,2-hydrogen-abstraction along with the cleavage of C-N bond can
give product P1 (NH3+CO) via the transition state TSRP1. While
CHONH2 also can degradation reaction by other two channels with
the barrier TSRP2, TSRa and TSab. In view of the four barriers, it is
impossible to overcome the barrier height for these four transition
states which the energy barrier is too high. According to the PES in
Figure 1a, there are two one-step and one two-steps degradation
reaction channels which are the possible reaction pathways. From
the kinetics, the pathway form P3 is more competitive. It needs to
overcome the barrier height 71.7 kcal/mol and 40.9 kcal/mol lower
than TSRP1 (80.0 kcal/mol) and TSRP2 (93.7 kcal/mol) about 8.3
kcal/mol and 22.0 kcal/mol. Meanwhile, the channel of forming P1
is one-step reaction pathway which is more feasible than two-steps
reaction pathway. While from the thermodynamics, the energy of
product P1 (5.4 kcal/mol) is lower than P2 and P3 by 10.2 kcal/
mol and 25.4 kcal/mol, respectively. Obviously, the formation of P1
is more favorable whatever in kinetics and in thermodynamics. The
degradation reaction channel can be described as:
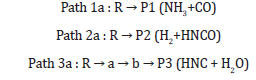
While from the real condition, the most feasible reaction
channel is hard to occur and degrade. How to degrade formamide
in industrial wastewater has been one of the problems for
environmental workers. In this work, we used the Au cluster to
catalysis the CHONH2 degradation reaction. Seen from (Figure
1), and compared to the CHONH2 degradation reaction, the
CHONH2+ Aux (x=0, 1, 2, 3) reaction mechanism is the same and the
formation of P1 is all more favorable whatever in kinetics and in
thermodynamics. The energy barrier of the transition state TSRP1
is 80.0 kcal/mol, 31.6 kcal/mol, 23.9 kcal/mol and 55.0 kcal/mol
respectively which catalyzed by Aux (x=0, 1, 2, 3). From a kinetic
perspective, the barrier of degradation reaction is the lowest when
x =2. On the other hand, from the thermodynamic perspective, the
degradation reaction can give off the most heat when x=1. It is
shown that the energy variation of the transition state is consistent
with the DE variation. However, because the inconsistency between
kinetic and thermodynamic conclusions, it is difficult to determine
which is the probable value of x in the formamide degradation
reaction catalyzed by Aux (x=0, 1, 2, 3) at different temperature
solely based on energies. To provide the exact x value of formamide
degradation reaction catalyzed by Aux (x=0, 1, 2, 3), there is need to
perform kinetic calculations.
Figure 1: A schematic potential energy surface of the CHONH2 degradation reaction obtained at the CCSD(T)//B3PW91 +
ZPE level.
In summary, starting from Reactant N H2CHO, we have found
three possible reaction channels (Paths 1~3). All the intermediates
and transition states of paths (Paths 1~3) proceeding lie above the
reactant R at the CCSD(T)//B3PW91 level, therefore, (Paths 1~3)
leading to P1 (NH3+CO), P2 (f + HNCO), and P3 (HNC + H2O) are much
less competitive at normal temperature. Of these three unfeasible
paths, we used the Au cluster to catalysis the CHONH2 degradation
reaction. We found Path 1 which forms product P1 (NH3+CO) is all
more favorable whatever in kinetics and in thermodynamics. The
relative energy of TSRP1 which catalyzed by Aux (x=0, 1, 2, and 3)
cluster lies 80.0 kcal/mol, 31.6 kcal/mol, 30.9 kcal/mol, and 55.0
kcal/mol; while thermodynamically, the formation of P1 is more
favorable by 5.4 kcal/mol, -23.8 kcal/mol, -8.5 kcal/mol, and -20.7
kcal/mol. Thus, we predict that the actual yield of the product
P1 may depend on the catalyst in the experiment. Therefore, as
reflected in the final product distributions, we predict that (1) a
total of three kinds of products P1 (NH3+CO), P2 (H2+HNCO), and P3
(HNC + H2O) should be observed; (2) the major product should be
P1 (NH3+CO), while P2 (H2+HNCO) and P3 (HNC + H2O) are the less
competitive products; (3) from the thermodynamic perspective,
the degradation reaction can give off the most heat when x=1; (4)
from a kinetic perspective, the barrier of degradation reaction is the
most lowest when the x is equal to 2. Since there is lack of product
information in experiment, we hope our present calculation may
represent a useful model for understanding the mechanism and
provide valuable information for further identification of the
product distributions for the title reaction, which is experimentally
unknown.
NBO Analysis
The natural bond orbital (NBO) were calculated in order
to understand various second-order interactions between the
filled orbital of one subsystem and vacant orbital of another
subsystem, which is a measure of the intermolecular delocalization
or hyperconjugation. NBO analysis provides the most accurate
possible “natural Lewis structure” picture of “j” because all orbital
details are mathematically chosen to include the highest possible
percentage of the electron density. A useful aspect of the NBO
method is that it gives information about interactions of both
filled and virtual orbital spaces that could enhance the analysis of
intra-inter molecular interactions. The second-order Fock-matrix
was carried out to evaluate the donor acceptor interactions in the
NBO basis. The interactions result in a loss of occupancy from the
localized NBO of the idealized Lewis structure into an empty non-
Lewis orbital. For each donor (i) and acceptor (j) the stabilization
energy (E2) associated with the delocalization i/j is determined as:
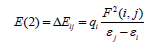
where qi denotes the occupancy of donor orbital, F(i, j)represents
the off-diagonal NBO Fock matrix element and ℇi and ℇj are orbital
energies (diagonal elements), respectively. In NBO analysis large
E(2) value shows the intensive interaction between electron donors
and electron-acceptors and greater the extent of conjugation of the
whole system, the possible intensive interactions are given in Table
2. The second-order perturbation theory analysis of Fock matrix
in NBO basis shows strong intramolecular hyper-conjugative
interactions of p electrons.

In Table 2, the perturbation energies of significant donor
acceptor interactions are present. (1) In CHONH2 , the interactions
between the LP (1) N4 and the anti-bonding of C1-O3 have the
highest E(2) value around 60.14 kJ/mol; (2) In CHONH2 +Au1, the
interactions between the LP (1) N4 and the anti-bonding of C1 have
the highest E(2) value around 86.90 kJ/mol. The other significant
interactions giving stronger stabilization energy value of 86.31 kJ/
mol to the structure are the interactions between LP (1) O3 and LP*
(1) C1; (3) In CHONH2 +Au2, the interactions between the LP (2) O3
and the anti-bonding of C1–N4 have the highest E(2) value around
17.08 kJ/mol; (4) In CHONH2 +Au3, the interactions between the LP
(1) N6 and the anti-bonding of C4 have the highest E(2) value around
86.23 kJ/mol. The other significant interactions giving stronger
stabilization energy value of 79.98 kJ/mol to the structure are the
interactions between LP (3) O9 and LP* (1) C4. Simultaneously,
substantial overlaps (Figure 2) of interaction orbitals are reduced
along with the decrements of E(2)s. The intermolecular electron
transfer occurs from the lone pairs of oxygen atom to the antibonding
orbitals of C–N bond, which can well explain the breaking
of C–N bond easily. Comparing with C–H and N–H bonds et.al,
partial orbital interactions between C and N in CHONH2 +Au2 is
weaker. Overall, the E(2)s in Table 2 indicates that the C–N bond in
CHONH2+Au2 is weaker than those in CHONH2 + Aux (x=0, 1, 3) to
some degree. This result is well consistent with kinetic parameters
obtained among reaction Path 1 in above reaction potential energy
surface analyses (Figure 3).

Conclusion
We have presented a detailed investigation on the related
energy and multi-channel CHONH2 degradation reaction potential
energy surface under the catalysis of Aux (x=0, 1, 2, 3) cluster and
its dynamic characterization using a detailed quantum chemical
methods. The following important conclusions are obtained
a) The prediction of CHONH2 degradation reaction under
the catalysis of Aux (x=0, 1, 2, 3) cluster with large positive
formation enthalpy hopefully will inspire and guide catalysis
efforts in this direction. NBO theory is applied to characterize
the nature of the intermolecular orbital interactions in the
CHONH2 + A ux (x=0, 1, 2, 3). The calculation show that the
maximum formation energy and thermodynamics prove that
the CHONH2 + Au1 reaction is the most possible, while kinetic
and NBO analyses prove that reaction CHONH2 + Au2 is the most
plausible. In summary, the rate constant of CHONH2 +Au2 is the
biggest one which is suitable with the kinetic and NBO analysis.
b) The multi-channel CHONH2 degradation reaction potential
energy surface under the catalysis of Aux (x=0, 1, 2, 3) cluster is
performed to explore the reaction mechanism. Our calculation
show that one primary channel is obtained which the H-shift
reaction can give the main product P1 (NH3+CO+ Aux (x=0, 1, 2,
3)). And CHONH2 + Aux (x=0, 1, 2, 3) reaction occurs mainly in
the high-temperature range which plays a more important role
as the temperature increase. The present theoretical studies
may provide useful information on the reaction mechanism.
c) The dynamic characterization show that the theoretical
rate constants are slightly positive temperature dependence
at 200~2000 K. The rate constant k of this reaction increases
significantly as the temperature increase. The analyses
consistently support the notion that the formamide degradation
reaction can be catalyzed by singlet atom whatever point, line
and surface in catalysts. Until now, there are no experimental
studies available to verify our results, but we hope to motivate
experimentalists to measure the rate constant.
Social Support and its Impact on Self- Sabotaging
Behavior: A Case Study - https://biomedres01.blogspot.com/2020/02/social-support-and-its-impact-on-self.html
More BJSTR Articles : https://biomedres01.blogspot.com




No comments:
Post a Comment
Note: Only a member of this blog may post a comment.