Targeting Hyaluronan in Vascular Diseases with Special Reference to Diabetic Macroangiopathy
Introduction
Vascular Extracellular Matrix (ECM) is a dynamic structure, which regulates blood vessel morphogenesis, provides organizational stability, and modulates various aspects of cell behavior [1]. Hyaluronan (HA) represents one of the most abundant macromolecules of the vascular wall. One of its main functions is to maintain blood vessel integrity [2]. Thus, any alteration in the metabolism of HA can predispose to the development and progression of atherosclerotic vascular diseases including diabetic macroangiopathy [3,4].
HA is a multifunctional linear glycosaminoglycan, composed of repeating disaccharide units of D-Glucuronic Acid (GlcUA) and N-Acetylglucosamine (GlcNac) (Figure 1). It is synthesized by specific plasma membrane glycosyltransferases, namely Hyaluronan Synthases 1-3 (HAS 1-3) [5]. The HASes exhibit distinctive enzymatic properties and use the cytosolic pool of UDP-sugars as substrates. The functions of HA are versatile and partly depend on its molecular size. For example, High Molecular Weight (HMW) HA possesses anti-inflammatory and immunosuppressive properties, whereas Low Molecular Weight (LMW) HA is a potent pro-inflammatory molecule [6]. The action of HA is also greatly influenced by its interactions with other ECM macromolecules such as lecticans, and cell membrane receptors, e.g. Cluster Determinant 44 (CD44) [7,8]. The turnover of HA is tissue specific and its half-life varies from 12 to 72 hours [9]. While HASes are responsible for the synthesis of HA, Hyaluronidases (HYALs) are enzymes responsible for its degradation.
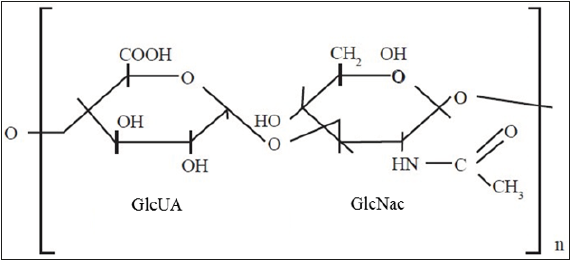
Figure 1: Schematic structure of HA. HA is an unsulfated, linear disaccharide composed of repeated D-Glucuronic Acid (GlcUA) and N-Acetylglucosamine (GlcNac) moieties. Note that HA is a glycosaminoglycan without any proteoglycan core protein. Modified from [30].
HA in Vascular Wall
HA is expressed in every type of blood vessel, and for example in arteries it can be detected in all three layers, mostly in the intima and adventitia and also in small amounts in the media. By maintaining intact endothelial glycocalyx, HA displays a pivotal role in vascular biology [10,11]. Overexpression of HA is characteristic for diabetic macroangiopathy [12]. Accumulation of HA occurs in response to hyperglycemia (Figure 2). This results in enhanced Vascular Smooth Muscle Cell (VSMC) proliferation and migration, and altered inflammatory cell adhesion properties [3,13]. Also increased HYAL activity and generation of Reactive Oxygen Species (ROS) are typical in diabetes. These both lead to HA fragmentation, which damages endothelial glycocalyx and thereby compromises vascular integrity even further [2,14]. Overall, dysregulation of HA turnover initiates processes that lead to various vascular complications including micro- and macroangiopathy in diabetic patients [15]. Thus, approaches targeting HA offer promising strategies (Figure 3) for novel therapeutic interventions in atherosclerotic vascular diseases.

Figure 2: HA-rich pericellular matrix of VSMCs. A; Cells grown in low glucose = normal physiological glucose concentration (5 mM). B; Cells grown in high glucose medium (25 mm) mimicking diabetic condition. Pericellular matrices are indicated by arrows in both panels. Scale bar = 100 μm. Note that hyperglycemia markedly increases the formation of HA matrices. Modified from [31].
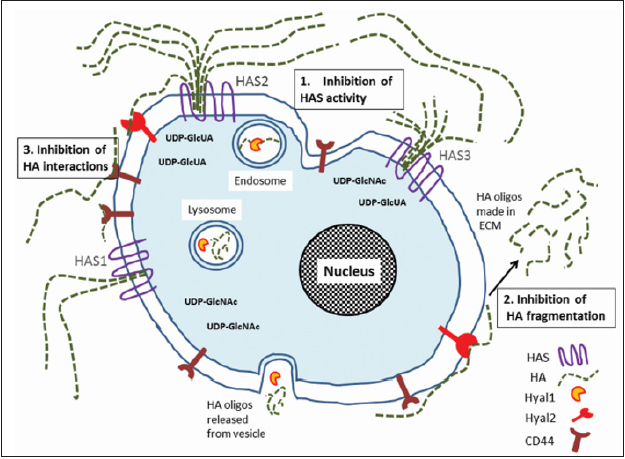
Figure 3: Targeting of HA in atherosclerotic vascular diseases. Novel therapeutic approaches are numbered 1-3 in the figure. 1; Accumulation of HA can be reduced by inhibiting the activity of HASes, particularly HAS2. 2; Decreasing the formation of pro-inflammatory LMW HA fragments. 3; Blocking of HA mediated signalling pathways by interfering interactions of HA with its receptors, e.g. CD44. Note that HASes produce different HA chain lengths (HAS1-2; 2x106 Da and HAS3; 2x105 Da) at different rates.
Targeting HA in Atherosclerotic Vascular Diseases
Inhibition of HA Synthesis
In diabetic macroangiopathy, the accumulation of HA has been recognized as the key event in the progression of atherosclerosis [12]. Atherosclerosis itself is an inflammatory disease, where the dominant hyaluronan synthase isoform expressing HA is HAS2 [10,13]. Thus, modulation of HAS2 activity provides an intriguing target molecule for therapeutic interventions in various vascular diseases [13,16].
Studies have been conducted to explore the potential value of 4-Methylumbelliferone (4-MU), a non-specific inhibitor of HA synthesis [17]. Unfortunately, systemic delivery of 4-MU has resulted in partial loss of the vascular glycocalyx, subsequently increasing inflammation. Specifically, 4-MU has been shown to increase recruitment of macrophages to vascular lesions and to facilitate progression of atherosclerosis [17].
Vascular inflammation is also coupled with the production of inflammatory cytokines, and for instance Interleukin-1β (IL-1β) is able to increase HA synthesis [10,18]. This offers a possibility to reduce vascular inflammation with anti-inflammatory treatments. For example, Has3 has been shown to be regulated by IL-1β [19]. Therefore, anti-IL-1β antibodies would block the activity of HAS3. Interestingly, deletion of Has3 gene has previously been shown to prevent VSMC-mediated neointimal hyperplasia in experimental vascular injury model [20]. However, deletion of Has2 is lethal already in the embryonic state.
HA production can also be influenced by the availability of HAS substrates, namely the cytosolic UDP-sugar pool [7]. The UDP-sugars are formed from glucose via hexosamine biosynthetic pathway [10]. Interestingly, a widely used diabetic drug metformin is able to downregulate the UDP-sugar pool of human VSMCs, thus reducing the provided substrates for HASes, and ultimately leading to decreased production of HA (Sainio et al., unpublished results). Indeed, metformin has been shown to elicit protective effect on cardiovascular events beyond its glucose-lowering effects [21]. Furthermore, the mechanism behind the inhibitory effect of HA synthesis by 4-MU mentioned above has also been linked to reduction of UDP-GlcUA levels [22]. In addition to the regulation of HAS substrates, it has also been suggested that HA synthesis could be modulated at post-translational level by regulation of the stabilization of the synthase proteins [23].
Inhibition of HA Fragmentation
Enhanced synthesis of HA is coupled with increase in HA catabolism during vascular inflammation [16]. This produces short HA fragments with pro-inflammatory properties. Also shedding of the vascular wall glycocalyx in diabetic patients has been shown to increase the amount of LMW HA, thus inducing inflammation [3]. Concomitantly, the vascular permeability is compromised. Fragmentation of HA can occur both via hyaluronidases and in a non-enzymatic fashion [24,25]. For example, reservation of the glycocalyx by inhibiting the function of HYAL1, the main somatic hyaluronidase, has been suggested as a therapeutic approach against diabetic vascular complications [11]. In addition to HYAL1, also HYAL2 and a recently found new hyaluronidase, TMEM2, degrade HA [23]. TMEM2 is located on cell surface and it is able to function also in higher pH than HYAL2. ROS-induced HA fragmentation represents an example of non-enzymatic process.
Modulation of HA Interactions
The best-known receptor for HA is CD44. The pro-inflammatory HA oligosaccharides interact with CD44 and another receptor group, the Toll Like Receptors (TLRs), stimulating inflammation processes [14]. Blocking of these interactions would attenuate inflammation and alleviate various vascular pathologies. This could be achieved for example with anti-CD44 antibodies, e.g. IM7, or A6- peptide [26]. Indeed, A6-peptide has been shown to block retinal vascular permeability in diabetes [27]. HA-receptor interaction can also be blocked with HA-binding peptides such as Pep-1 [28]. Furthermore, use of HA as a tool to interfere signaling pathways of other inflammatory ECM macromolecules would provide a potential therapeutic target. This has been seen in chondrocytes, where biglycan induced inflammation has been demonstrated to be attenuated by administration of HMW HA [29].
Conclusion
As discussed above, HA is a vital regulator of vascular integrity and dysregulation of its synthesis and turnover can lead to pathologies such as diabetic macroangiopathy. Because of this, targeting HA synthesis, prevention of HA degradation and inference with HA interactions would provide potent therapeutic approaches. Also, novel therapies targeting the degradation of accumulated HA waits to be discovered.
The Therapy of Cancer-https://biomedres01.blogspot.com/2020/11/the-therapy-of-cancer.html
More BJSTR Articles : https://biomedres01.blogspot.com



No comments:
Post a Comment
Note: Only a member of this blog may post a comment.