Generation of Acsl4 Gene Knockout Mouse Model by CRISPR/Cas9-Mediated Genome Engineering
Introduction
Porcine intramuscular fat (IMF) content is considered one of the most important traits of pork quality, and has been positively correlated with meat tenderness, moisture content and taste [1]. Therefore, it is necessary to search for new candidate genes and molecular mechanisms associated with IMF deposition. The gene coding for Acyl-CoA synthetase long chain family 4 (Acsl4) plays an important role in lipid synthesis and fatty acid degradation [2]. The porcine Acsl4 gene is located on chromosome X, close to a quantitative locus for IMF [3]. Chen (2014) reported that pig Acsl4 gene seems to be a candidate gene to improve IMF content in pig breeding system, because of the association of the RsaI polymorphism and IMF in 2 native Chinese pig breeds(Yanan and Jinhua pigs) and DLY (Duroc x(Landrace x Yorkshine) pigs [4]. All these studies suggest that Acsl4 is an ideal target for increasing IMF in livestock, but the detail mechanism is still not clear. As we known, the disruption of genes as knockout approaches has provided a powerful tool for characterizing gene function. Hence, the generation of Acsl4 knockout animal model is of great significance for investigating its effects on intramuscular fat deposition. Animal model with targeted gene deleted provide us important tool for gene function analysis, while it is very difficult for us to produce a geneknockout pig model because of the costs associated with housing and maintaining large animals and long generation interval, even if the CRISPR/Cas9 system has been widely used and had greatly simplified the program.
So it is expected to create a mouse model with Acsl4 gene deleted, and there has been no report of large deletions of Acsl4 gene in mice. Clustered regularly interspaced short palindromic repeats/CRISPR-associated systems (CRISPR-Cas) has emerged as a powerful tool for animal model generating due to its high efficiency and easy to operate in recent years. Many genetically modified animals, such as zebrafish [5], Drosophila [6], mouse [7] and pig [8] have been generated using the CRISPR/Cas9 system. When only one sgRNA was injected, small insertions or deletions less than 100bp were typically produced, and further breeding and genetic testing of the homozygous offspring were therefore necessary. Using dual sgRNA injection system could create large gene deletions [9,10]. Here, we report the use of dual sgRNA-guided CRISPR/Cas9 system to generate Acsl4 knockout mutants in mice. Co-injection of mice embryo with two sgRNAs results in the elimination of an interval of 12145 bp between two guide RNAs. It is the first Acsl4 knockout (KO) mouse model to allow the functional analysis of Acsl4 gene in mammalian vertebrate, and it also can be applied for screening lipid metabolism related genes.
Methods
Ethics Statement
All animal studies were approved by the Animal Welfare and Research Ethics Committee at Hubei Academy of Agricultural Science, and all procedures were conducted strictly in accordance with the Guide for the Care and Use of Laboratory Animals. All surgery was performed under anesthesia, and every effort was made to minimize animal suffering.
Design of Guide RNA
The online software package ZiFiT Targeter version 4.2 (http:// zifit.partners.org/) was used to identify guide RNA (gRNA) target sites requiring the structure GG-(N)18-NGG [11]. The targets were selected for target exons of mouse Acsl4. The gRNA 1 and gRNA 2 sites spanning 5 exons were chosen to delete large DNA fragments. To construct gRNA expression vectors, gRNAs, sense and anti-sense oligomers of the gRNAs were synthesized. A 5’-overhang TA- or AAAC-was included in each gRNA oligomer were allow directional cloning into an expression vector.
Production of Guide RNAs and Cas9 mRNA
To construct the gRNA expression vector, pDR274 (Addgene ID:42250) was digested with the BsaI restriction enzyme . And then, a pair of gRNAs encoding the 20-nt guide sequences was annealed at 95 ℃ for 5min and ramped down to 25 ℃ to generate the dsDNA fragment, which was then cloned into the BbsI-digested pDR274 vector. PCR products were amplified with T7-F (5’-TAATACGACTCACTATAG-3’) and T7-R(5’- AGCACCGACTCGGTGCCACTT-3’) primers using the constructed gRNA expression pDR274 vector as templates. After that, the sgRNAs were in vitro transcribed with the T7 RNA Synthesis Kit (Ambion) using the PCR products amplified according to the manufacturer. The concentration and quality of synthesized mRNAs were determined by Nanodrop 2000 and agarose gel electrophoresis. The Cas9 gene expression vector was ordered from Addgene (Addgene plasmid #42251) and linearized with PmeI and transcribed in vitro using the mMessage mMachine SP6 Kit (Life Technologies).
Microinjection and Embryo Transfer
The protocol for microinjection of pronuclear-stage embryos has been described in detail by our published protocols [12]. In brief, zygotes were collected from sexually immature female C57BL/6 mice (4-5 weeks old), that had undergone superovulation by intraperitoneal injection of PMSG and hCG and mated overnight with C57BL/6 male mice that over 12 weeks old. The oviducts were then flushed and pronuclear-stage embryos were transferred to embryo culture medium for microinjection. Mixtures of gRNA (25ng/μL) and Cas9 mRNA (100ng/μL) were injected into the cytoplasm of pronuclear stage embryos. The injected embryos were transferred to embryos culture medium for 30-60min, followed by transfer of approximately 15-20 injected embryos into the oviduct of the recipient mother for KO mouse productions. The founders were genotyped by PCR followed by DNA sequencing analysis. The positive founders were bred to the next generation (F1) and subsequently genotyped by PCR and DNA sequencing analysis.
Genotyping Analysis
Genomic DNA was extracted from the tails of mice and analyzed via PCR using primers: Acsl4- F (5’-GCTAAGCCCACTTCAGACAAACCTG- 3’) and Acsl4-R (5’-ATGGTCTTCCCCATGATGCTAACAC- 3’).The reaction program comprised the following steps: 95 ℃ for 2min, 40 cycles of 94 ℃ for 30s, 56 ℃ for 30s and 72 ℃ for 30s, followed by 72 ℃ 5min. The PCR products were gel purified and cloned into the pMD18-T vector (Takara, Dalian, China) and sequenced to determine the exact sequences of both alleles. The DNA sequencing primer (Reverse Sequencing) is: 5’-TTCCAGCACCACATGATTCTGTC- 3’.
Reverse Transcription PCR Analysis of Acsl4 mRNA: Total RNA was extracted using the TRIzol reagent (Life Technologies), and the RNA was quantified with a Nanodrop spectrophotometer (Thermo Scientific).First-strand cDNA was synthesized from total RNA (1μg) with a SuperScript III Cells Direct cDNA Synthesis Kit (Invitrogen). The Acsl4 cDNA was amplified with real-time PCR, using the primers Acsl4-C-F(5’-GCCCACTTCAGACAAACCTGG-3’) and Acsl4-C-R(5’-ACAGCTTCTCTGCCAAGTGTGG-3’). GAPDH cDNA was amplified with PCR, using the primers GAPDH-C-F (5’-GGTGAAGGTCGGTGTGAACG- 3’) and GAPDH-C-R (5’-CTCGCTCCTGGAAGATGGTG- 3’). The reaction conditions were as follows: 95 ℃, 5min; 35 cycles of 95 ℃ for 30sec, 56 ℃ for 30sec, and 72 ℃ for 30sec; and then 72 ℃, 5min. PCR produce size was estimated by electrophoresis of 5μL aliquots on a 2% agarose gel.
Results
Construction of CRISPR
Acsl4 protein is a master regulator of lipid metabolism. Mutations in the pig Acsl4 gene have been found in pigs with high IMF content, its prompts us to generate Acsl4 gene knockout mouse to model this phenomenon. The mouse Acsl4 gene (Genbank accession number:NM_207625.2) is located on mouse chromosome X. Sixteen exons have been identified, with the ATG start codon in exon 3 and TAA stop codon in exon 16. Expn 5 to exon 10 was selected as target sites. We designed two sgRNAs targeted on exon 5 and exon 10 of the mouse Acsl4 gene (Figure 1) The sgRNA target sequence did not cross-react with any other sites in the mouse genome and was followed by an NGG protospacer adjacent motif Copyright@ Hongyan Ren| Biomed J Sci & Tech Res| BJSTR. MS.ID.002506. 3/5 (PAM), which is necessary for Cas9 cleavage. The sequences of gRNA are listed in (Table 1). Cas9 mRNA was generated by the in vitro transcription of a linearized and T7 promoter-driven plasmid template, which included a human-codon-optimized version of Cas9 cDNA and nuclear localization signals (NLS) at 3’ end of Cas9. Good-quality purified sgRNAs and Cas9 mRNA were used for oocyte injection (Figure 2).
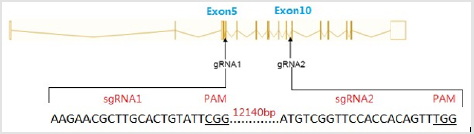
Figure 1: Schematic representation of sgRNAs specific to exon5 and exon 10 of the mouse Acsl4 locus.
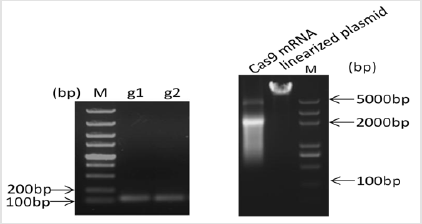
Figure 2: Design and construction of CRISPR. The quality of sgRNA and Cas9 mRNA was assessed by gel electrophoresis. sgRNA showed as a single band nearly 100bp; Cas9 mRNA showed as 2 bands due to secondary structure.

Table 1: The sequences of gRNAs.
Generation of Acsl4 Knockout Mouse Model
The Cas9 mRNA and two sgRNA were co-injected into the cytoplasm of mouse zygotes. The tail of the newborn mice were dissected, PCR and real-time PCR were performed to confirm the disruption of ACSL4 in these mouse (Table 2). Compared with wild-type mouse, the desired large fragment deletion of Acsl4 gene spanning both the sgRNA1 and 2 sites were detected in 14#,18# and 19# (Figure 3). The T-cloning and sequencing results demonstrate that a large deletion about 12145bp can be achieved via the dual gRNA system (Figure 4). Determination of 12145bp deletions of Acsl4 gene in mice by PCR. A Clear band demonstrates the largefragment deletion of Acsl4 in 14#,18# and 19# mice. WT: wild type control; The expression level of Acsl4 mRNA was further examined by quantitative real-time PCR in the skin tissues of tail from wild type and mutant mouse, the results confirmed that the Acsl4 mRNA level were lower in the mutant mice than the WT mice (Figure 5).
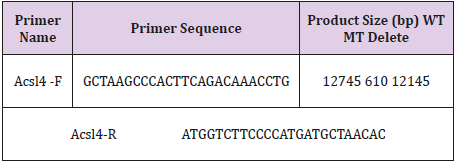
Table 2: The primers and conditions for PCR genotyping.
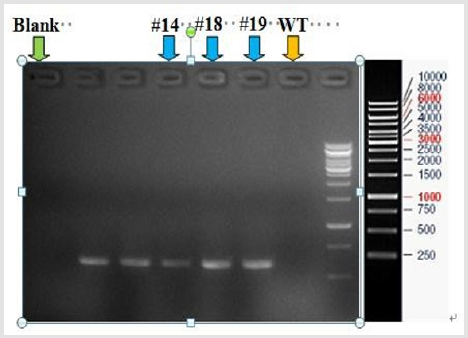
Figure 3: Dual sgRNA-induced large deletion at the mouse Acsl4 locus.
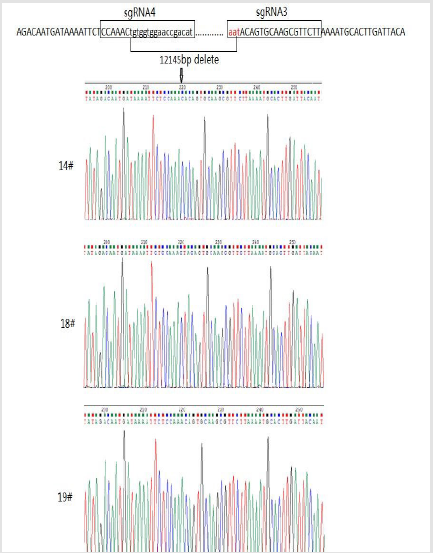
Figure 4: Genomic sequences of CRISPR target regions in mutant mice.
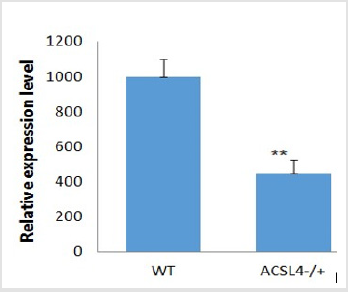
Figure 5: Real-time PCR for Acsl4 mRNA expression analysis.
Discussion
Loss-of-function is the most frequently employed strategy to study a target gene in molecular biology. Genetic knockout of gene is the most reliable technique of study of loss-of-function of gene with high efficiency and specificity for in vivo analysis; however, the manipulation procedure is complicated and the process is time consuming. Therefore, to establish a robust and convenient technique in study of gene loss-of-function is in need. Targeted mutations to the gene Acsl4 mediated by the CRISPR/ Cas9 system have not been reported in mice previously. In this report, we described the feasibility of applying the Cas9/gRNA system to produce a large deletion of the Acsl4 gene in mice. To our knowledge, this is the first description of large fragment deletion of Acsl4 gene in mice. The CRISPR/Cas9 system has emerged as a new genome editing technology in mice. In principle, this approach is limited only by the requirement of a PAM sequence [13]. Here, we report a simple and efficient method to generate gene knockout mutants in mice using the CRISPR/Cas9 system. Specifically, we showed that the co-injection of two sgRNAs into mice can induce a deletion of the chromosome sequence between the two sgRNAtargeted loci, which is useful for creating null alleles.
In addition, we showed that large deletions can be easily identified by PCR and agarose electrophoresis, which is especially important for those mutants with no visible phenotypes. Encoding multiple guide sequences into a single CRISPR experiment to simultaneously edit several sites within the genome has been used to study gene function in mammals [14]. In addition, the simultaneous use of multiple sgRNAs to target an individual gene has been reported to improve genome-editing efficiency. Our data suggest that multiplexed guide RNAs may cause the off-target deletion of chromosome sequences in addition to gene editing at the targeted loci. Therefore, independent cell clones and rescue experiment are required to characterized gene function in mammalian cells in this set of experiments. Our results suggest that dual sgRNAs are more efficient in inducing mutations than individual sgRNAs, and the CRISPR/Cas9 system is more efficient than other traditional gene editing tools [15,16]. The frequency of simultaneous cleavage by two sgRNAs was higher than expected. It is possible that this enhanced efficiency was partly due to homology-directed repair in which one mutant allele served as the template for the other allele. Alternatively, the DSBs induced by the first sgRNA may have increased the genomic accessibility of a nearby region, which could facilitate the cleavage of the second sgRNA.
The surveyor assay and T7E1 assay have been widely used to detect the mutation efficiency induced by custom endonucleases . Both methods largely depend on randomness and efficiency during re-annealing. On the other hand, several studies have specifically chosen restriction enzyme sites as targets to facilitate the detection of mutations. In this study, we evaluated the mutation efficiency by observing the chromatogram of PCR amplicons ,and the results were consistent with a subsequent TA cloning experiment. Therefore, it is convenient to roughly estimate the mutation efficiency by analyzing the height of multi-peaks in the chromatogram. Off-target effects are a major concern in the Cas9-mediated gene editing system [17,18]. Such effects were not found in the present study. We suspected that a low concentration of sgRNA and Cas9 mRNA would reduce off-target effects; furthermore, it is particularly important to avoid mismatches in the seed sequence, which are critical for site-specific cleavage in the CRISPR/Cas9 system.
Regional pH in Five Agricultural Soil-Types – Associations with Temperature and Groundwater Si in Continental Finland-https://biomedres01.blogspot.com/2021/02/regional-ph-in-five-agricultural-soil.html
More BJSTR Articles : https://biomedres01.blogspot.com



No comments:
Post a Comment
Note: Only a member of this blog may post a comment.