Immunodiagnostic Applications of Polyclonal Antibodies that Recognize the Southern Bean Mosaic Virus Movement Protein
Plant viruses invade and spread through their hosts either via the short-distance movement which involves the spreading of the virus from the infected cell to the adjacent cell, or, via the long-distance movement where the virus spreads through the vascular system and infects cells from different regions of the plant. In the first case, viruses penetrate the cell wall through the plasmodesmata, which are channels unique in the plant kingdom, typically with diameters of about 25-30 nm, that interconnect cells [1-3]. Different species of plant viruses encode specialized proteins referred to as Movement Proteins (MPs) that dilate the plasmodesmata permitting the passage of complexes formed by viral nucleic acids and proteins or even of whole virions [4].
There is no common movement mechanism that is shared by all viruses and viruses, independent of their taxonomy, may resort to different strategies depending on the MP sequence, participation of virus encoded proteins, type of infected host and interaction with host factors [5-7]. MPs, as well as replicases are non-structural proteins that are generally produced in very low concentration in plants thus making it difficult to identify, localize and purify them [8].
The aim of this study is the production of antibodies for the efficient detection of low concentrations of MPs from the Southern Bean Mosaic Virus (SBMV) which belongs to the Sobemovirus genus. This virus has a strict host range and some of them infect common bean and soy, which are of economic relevance. SBMV is the type species from this genus, has isometric particles (28- 30 nm) and contains genomic RNA of 4.0-4.5 Kb with positive polarity and a capsidal protein with a molecular mass of 29-30 kDa. The SBMV genome consists of four Open Reading Frames (ORFs) with sequence overlaps among them. ORF 1 encodes a movement protein with 17 kDa [9].
Materials and Methods
Virus Purification and RNA Extraction
SBMV was purified from infected leaves of Phaseolus vulgaris following the method of [10]. Viral RNA was extracted using the RNeasy Plant Mini Kit (Qiagen) following the manufacturer’s instructions.
Primer Design, cDNA Synthesis and PCR Amplification of the ORF1
Based on the sequence deposited with the GenBank database under Accession No. DQ875594.2, primers were designed as shown below to amplify the ORF1 from genomic RNA of an isolate of SBMV. BamHI and Hind III restriction sites were incorporated into the forward and reverse primers, respectively.
Forward primer: 5’ – CCGGATCCATGAGCTATCGT – 3’ (BamHI restriction site highlighted).
Reverse primer: 5’ – AAGCTTCCGCGCAGATAA – 3’ HindIII (XhoI restriction site highlighted).
First strand cDNA was synthesized using Superscript II Reverse Transcriptase (Invitrogen), and used as a template for the Polymerase Chain Reaction(PCR) for amplification of ORF 1 performed in 100 µl mixture containing 50 ng of genomic RNA, 1µL (10 µM) of each primer, 2 µl (10 mM) dNTPs, 10 µl of PCR buffer (100 mM Tris–HCl pH 8.8, 500 mM KCl, 0.8% 25 mM MgCl2), and 2.5 U of native Taq DNA polymerase enzyme (MBI Fermentas). The amplification was carried out using the following reaction cycles: initial denaturation at 95 °C for 10 min followed by 30 consecutive cycles of denaturation at 95°C for 30 s, annealing for 1min at 55°C, extension at 72°C for 1 min 30s, then final extension at 72°C for 10 min. Electrophoresis on a 1% agarose was used to confirm the specific PCR product obtained and it was purified using PCR gel purification kit (Qiagen).
Cloning in pGEM-T Easy Vector
The resulting PCR product and pGEM-T Easy vector were ligated at 4°C overnight using T4 DNA ligase to yield the pGEM-TMP construct that was transformed into Escherichia coli DH5αcells. To confirm successful cloning, blue white screening colony and PCR was performed. One clone was used for plasmid extraction and the recombinant vector was sequenced by the automatic ABI 377 DNA Sequencer.
Subcloning in pET28a Vector and pMALc2x
Recombinant pGEM-T-MP vector was digested with BamHI and HindIII restriction enzymes and a 500bp insert (ORF) was purified from agarose 1% gel by using gel extraction and purification kit (Qiagen). This product was cloned in pET28a vector (Novagen) and pMALc2x (New England Biolabs) yielding pET28a-MP and pMALMP respectively. Positive clones were first selected by PCR and reconfirmed by restriction digestion. Using pMALc2xsystemthe predicted molecular weight of recombinant MP isapproximately 60 kDA, 42.5 kDa of Maltose Binding Protein (MBP) + 17 kDa of MP and using pET28a is approximately 20 kDa, 17 kDa of MP + His tag followed by the polylinker sequence.
Expression of Recombinant Protein in E. coli using the pMALc2x Vector
For the expression of the MP protein tagged with MBP, BL21 E.coli cells were transformed with the pMAL-MP construct and incubated in LB broth (with 0.2% glucose and 100 µg/ml ampicillin for selection). The medium was inoculated with an overnight culture (1:100 dilution) and the culture was incubated under constant agitation at 25°C until an OD6000.5 was reached. Subsequently, 0.3 mM IPTG was added and the culture was further incubated either at 37°C or at 25°C for 6 hours.
Purification of the MP-MBP Recombinant Protein
Cells were harvested by centrifugation at 6,000 x g, at 4°C for 20 min and lysed in a buffer containing 20 mM Tris-HCl pH 7.4, 200 mM NaCl, 1 mM EDTA (including protease inhibitors: 100 µg/ ml PMSF, 0.5 µg/ml leupeptin, 0.5 µg/ml aprotinin and 1 µg/ml pepstatin) by sonication for 2 min on ice. The crude extract was isolated by centrifugation of the samples at 15,000 x g at 4°C for 1 hour and was applied on to a column containing an amylose resin pre-equilibrated with the lysis buffer. The recombinant protein was eluted after the addition of the lysis buffer with 10 mM maltose and a final step of gel filtration using a Superdex G75 10/300 GL column, was performed. The presence of the MP through all the steps of expression and purification steps was monitored by SDSPAGE gels.
Preparation of Polyclonal Antibodies against the Recombinant MP
Antibodies against the recombinant MP were raised in a male New Zealand white rabbit which was injected subcutaneously with 1 mg of the purified recombinant MP-MBP protein in 5 mM Tris-HCl pH 7.5; 100 mM NaCl. Four booster injections were administered with 1 mg of recombinant protein each at an interval of one week. One week after the final injection, 50 ml of blood was collected and maintained overnight at room temperature. The crude antiserum was collected by centrifugation (4200 x g for 5 min) and the globulin fraction was isolated by three rounds of selective precipitation with ammonium sulfate (40% saturation). After the final round of precipitation, the proteins were dissolved in 20 ml of 5 mM Tris-HCl pH 7.5, 200 mM NaCl. The titer and the specificity of the antiserum were checked by western blot and dot blot.
Expression of Recombinant Protein in E. coli using the pET28a Vector
For expression of the His-tagged MP, BL21 E.coli cells were transformed with the pET28a-MP construct and incubated in LB broth (100 µg/ml kanamicin for selection). The medium was inoculated with an overnight culture (1:100 dilution) and the culture was incubated under constant agitation at 30°C until an OD600≌0.5 was reached. Subsequently, 0.3 mM IPTG was added and the culture was further incubated either at 30°C or at 18°C for 8 hours.
Purification of His-Tagged MP
Expression of MP using pET28a in E. coli was fraught with difficulties, including poor expression, degradation, and sequestration as insoluble protein in inclusion bodies; therefore it was purified under denaturing conditions and subsequently refolded. After sonication, the insoluble fraction from 1 liter of culture was washed 5 times with a wash solution (20 mM Tris-HCl pH 8.0, 50 mM NaCl, deoxycholate 2%, 1 mM DTT) and resuspended in 20 ml denaturing buffer (100 mM NaH2PO4 pH 8.0, 8 M urea). Cell debris was removed by centrifugation at 15,000 x g for 20 min. The recombinant MP was purified under denaturing conditions using Nickel Sepharose (GE). Initially the column was loaded with 20 ml of the binding buffer (100 mM NaH2PO4 pH 8.0, 8 M urea) and washed with 6 column volumes of the wash buffer (100 mM NaH2PO4 pH 6.3, 8 M urea). The column bound protein was subsequently eluted with 10 ml of elution buffer (100 mM NaH2PO4, pH 4.0, 8 M urea) and the concentration was determined, adjusted to 1 mg/ml and checked by SDS-PAGE gels. The purified protein was used in western blot, dot blot and refolding assays.
Refolding of MP
His-tagged MP was refolded following a simple dilution protocol in Tris-HCl pH 7.5 buffer which additionally contained 200 mM NaCl, 1 mM DTT , 1 M 3-(1-Pyridinio)-1-propanesulfonate, at 4oC, wherein the 8 M urea solution containing the recombinant MP (1 mg/ml) was diluted to a concentration 10 µg/ml. The sample containing the diluted purified protein was concentrated by centrifugation inan AMICON (3.000 Da) micro-concentrator by centrifugation at 4,000 x g at 10°C.
Circular Dichroism Spectroscopy of Refolded MP
Far UV-CD spectra in the range 190-260 nm were obtained with quartz cells with a path lengths of 0.5 mm and a scan rate 50 nm/min at 25oC with a response time of 1.0 second, spectral bandwidth of 1.0 nm and a spectral resolution of 0.1 nm by utilizing a Jasco J-710 Spectropolarimeter (Jasco, Tokyo, Japan). For each spectrum, 10 accumulations were performed. Secondary structure percentages for each tested condition were calculated with CONTINLL software of CDPro package, using the reference set of proteins SMP56 [11].
Western Blot Evaluation
To determine the specificity of polyclonal antiserum, MP Histagged that has no MBP (10 µg) was used in immunoassays to make sure that the polyclonal antibody has affinity for the sequence encoded by ORF1 avoiding reaction with MBP fusion protein. MP His-tagged, and two negative controls, the recombinant C-terminal portion of RNA polymerase of SBMV (500 µg) and BSA (500 µg), were applied on 15% SDS-PAGE gels, and then electroblotted onto a 0.22 mm pore size nitrocellulose membrane at a constant current of 80 mA for 1 h. An identical gel was run and stained with Comassie brilliant BlueR-250. After blocking in TBS (20 mM Tris-HCl pH 7.4, 150 mM NaCl) with 5% (w/v) skimmed milk, the membrane was incubated with polyclonal antibodies against MP-MBP (1:2000 diluted in TBS) at 25°C for 2 h. After washing three times, goat-antirabbit IgG conjugated with alkaline phosphatase (Sigma) was added and incubated for 1 h at room temperature. The immunoreactive bands were subsequently visualized by using 5-Bromo-4-Chloro3-Indolylphosphate (BCIP) and Nitro Blue Tetrazolium (NBT) as a substrate.
Western Blot Analysis of Proteins Extracted from Infected Bean Leaves
Following inoculation of bean leaves with the purified virus for different time intervals, the bean leaves were harvested, and 1 gram was homogenized by grinding in 1 ml of 50 mM Tris-HCl, pH 7.5, 9 M urea, 10% SDS, 5% 2-mercaptoethanol. The sample was boiled for 5 min, centrifuged at 10,000 x g for 10 min and 20 µL of the supernatant was applied in 12% SDS-PAGE gels. The proteins were electro-blotted onto a nitrocellulose membrane and submitted for Western blot assays as described above.
Dot Blot
The purified recombinant movement protein was also analyzed by dot blot in two different experiments. First, 20 ng of the Histagged MP was spotted onto strips of a nitrocellulose membrane and was used to determine the efficiency of the obtained antiserum that was diluted 2000 fold. The second experiment consisted of a serial dilution of the recombinant protein in PBS that was spotted on the membrane at 2 µl/spot and incubated with polyclonal antibodies against MP-MBP (1:2000 diluted in TBS) at 25°C for 1 h. The samples were washed and visualized following the Western blot analysis protocol. This experiment was repeated twice using both denatured and refolded proteins.
Prediction of Linear Epitopes
Putative linear epitopes of MP of SBMV were predicted using the program BepiPred [12]. The prediction score is based on hydrophilicity and secondary structure prediction from a single sequence.
Results and Discussions
Virus Purification, RNA extraction and Cloning of ORF1 from SBMV
From infected bean leaves it was possible to purify viral particles with high purity (Figure 1), serving as a source of RNA for cDNA synthesis. In this study, ORF1 of SBMV was cloned into the expression vectors pMALc2x and pET28a.
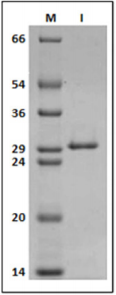
Figure 1: 12% SDS-PAGE gel labeled in kDa; (M) molecular marker, (1) Purified Southern bean mosaic virus.
Expression and Purification of Recombinant MP-MBP
Using pMALc2x it was possible to purify a recombinant movement protein of approximately 60 kDa (Figure 2). This expression vector was chosen not only based on solubility and stability of MBP-MP, but also because MBP might enhance immune responses to vaccine fusion proteins [13,14] facilitating the production of antibodies as it has been demonstrated in mice and rabbits eliciting a strong IgG antibody response against both the dengue virus [15,16] and Plasmodium falciparum [17].
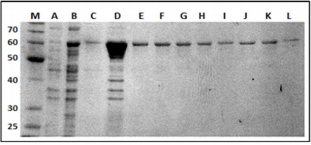
Figure 2: 12% SDS-PAGE gel of the purified MP-MBP labeled in kDa. Lane (M), molecular-weight markers; (A) non-induced E. coli sample; (B) E. coli sample 1 hour after IPTG induction;(C) following the wash step. Lanes D, E, F, G, H, I, J, K and L, represent elution fractions of MP-MBP.
SDS-PAGE analysis of the recombinant clone containing recombinant pMALc2x-MP indicated high levels of production of soluble recombinant proteins after induction with IPTG, permitting purification in the native state by conventional amylose resin chromatography. A final step of gel filtration utilizing a Superdex G75 10/300 (GE-Healthcare life sciences) column was performed. The protein was concentrated to 1mg/mL, the purity was checked by SDS-PAGE gels (Figure 2) and was used for the production of polyclonal antibodies in rabbits.
Expression and Purification of Recombinant Protein in E. coli using pET28a Vector
This system was successful in expressing the N-terminal Histagged MP that was subsequently purified under denaturing conditions.
Circular Dichroism Spectroscopy of Refolded MP
The refolding of MP was analyzed based on the percentages of secondary structure calculated by the CONTINLL software of the CDPro package, using the reference set of proteins SMP56 [11] and indicate a structural content of 25 % α-helix, 39 % β-sheet, 18 % turns and 18 % random coil (Figure 3). Using the purified recombinant MP-MBP as the antigen, its polyclonal antiserum was prepared successfully through the procedure described in the Materials and Methods. To avoid reactions among the antibodies and the Maltose Binding Fusion Protein (MBP) in immunoblotting assays, all experiments were performed using MP His-tagged, ensuring that during experiment the antibodies recognize specifically the amino acid sequence encoded by ORF1 of SBMV.
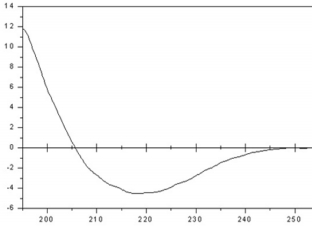
Figure 3: Circular dichroism spectrum of a sample of the refolded MP. The protein conformation consists of 25 % α-helix, 39 % β-sheet, 18 % turns and 18 % random coil.
Western Blot
Western blot showed the specificity of the antibodies reacting positively with MP His-tagged (20 kDa), but not with BSA and recombinant C-terminal portion of SBMV polymerase, even 5 times more concentrated than the MP (Figure 4).
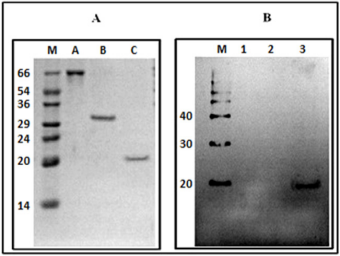
Figure 4: Panel (A) 12% SDS-PAGE gel (numbers indicate kDa); (M) molecular-weight markers; (A) BSA; (B) C-terminal domain of SBMV polymerase; (3) His-tagged SMBV MP. Panel (B) Western blot using the anti-histidine monoclonal antibody. (M), molecular marker, Magic Mark XP (Invitrogen) (1) BSA, (2) C-terminal portion of the polymerase of SBMV; (3) MP His-tagged of SBMV.
Dot Blot
Twenty ng of the refolded His-tagged MP spotted onto strips of nitrocellulose membrane was used to determine the efficiency of the obtained antibody and we found that the antiserum at different dilutions (1:1.000; 1:2.000; 1:4.000; 1:8.000; 1:16.000; 1:32.000) reacted with the recombinant protein. The same assay was performed using denatured and refolded MP and no difference was found between them (Figure 5). The minimal dilution of antiserum able to react with 20 ng of MP was 1:16000. Analyzing serial dilutions of recombinant MP His-tagged, it was possible to conclude that at least 1.8 ng of protein is detectable using the antiserum diluted 1:2000 (Figure 6).
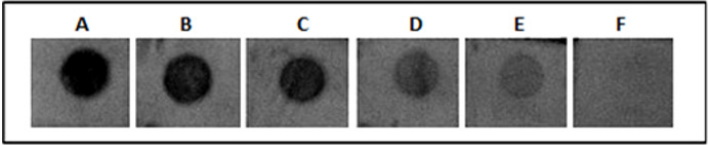
Figure 5: Titration assay by dot blot using polyclonal antibody against MP of SBMV. Each dot contains 20 ng of refolded MP His-tagged. A to F represent the following dilutions of antiserum: 1:1.000; 1:2.000; 1:4.000; 1:8.000; 1:16.000; 1:32.000.
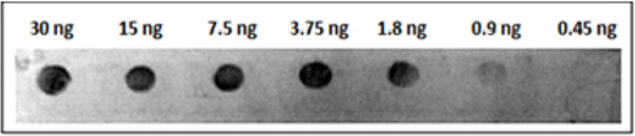
Figure 6: Titration assay by dot blot using polyclonal antibody against MP of SBMV. Different concentration of MP of SBMV were detectable using the antiserum diluted 1:2000.
MP of SBMV was not detected in experiments using proteins from the crude extracts of bean leaves indicating that the sample applied in membrane has MP concentration lower than 1.8 ng (90 ng/gram of sample of infected leaf) or that MP was degraded during the viral infection cycle.
Prediction of Linear Epitopes
The Bepipred Linear Epitope Prediction tool was used in order to explain the antibodies recognition of denaturing and native form of MP guided by algorithms that predict immunogenic epitopes. Four main linear epitopes were predicted (Figure 7). Antibodies may recognize epitopes made up of contiguous (conformational epitope) or non-contiguous (linear epitope) amino acids and according to these results, it is suggested that the MP has surfaceexposed linear epitopes in native state. The advantage of using an antiserum that recognizes both native and denatured proteins is the possibility to use additives that can change the folding of the target protein without disturbing its recognition.
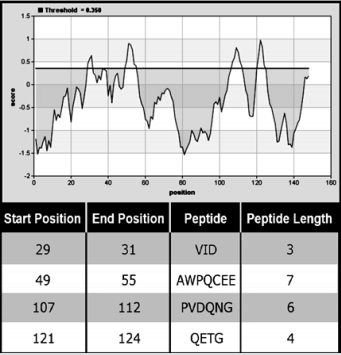
Figure 7: Graphical result of Bepipred Linear Epitope Prediction tool. The Positive scores for linear epitopes and their predicted epitopes along with each respective sequence are showed in the table associated with the graphic.
Conclusion
The movement protein was cloned and over expressed in E. coli and was subsequently used as an antigen to produce antibodies against the recombinant protein with high specificity. Since the prepared polyclonal antibody is able to react specifically with MP protein of SBMV under both denaturing and native conditions, it may serve as a tool to identify its sub-cellular localization and can help to understand its role in virus replication. Based on immunoblotting assays of infected bean leaves we conclude that the MP concentration per gram of sample of infected leaves is less than 90 ng or that the MP is degraded during the viral infection cycle.
Acknowledgement
This work was supported by grants from Conselho Nacional de Desenvolvimento Ciêntifico e Tecnológico (CNPq/Brazil) Grant 307338/2014-2 and FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo) Grant numbers 2018/10736-8; 2018/07977- 3; 2015/13765-0 and 2015/18868-2.
More BJSTR Articles : https://biomedres01.blogspot.com/



No comments:
Post a Comment
Note: Only a member of this blog may post a comment.