Nucleotide Variations in microRNA-Binding Sites: The Need of Novel Tools for the Nucleic Acid Alignment
Introduction
During the last fifteen years, the attention of biologists and
medical scientists has been attracted to microRNAs, the small
regulatory molecules of about 22 nucleotides long. The suppressive
effect of microRNAs on gene expression is well-known and
generally attributed to microRNA-mediated degradation of its
target messenger RNA (mRNA, the molecule produced by the gene),
messenger RNA translational repression, or both. To accomplish
this, microRNA aligns with its mRNA target upon the principles of
complementarity between the microRNA and mRNA nucleotides
[1] (Figure 1). It has been suggested that naturally-occurring
variations in microRNAs and their target genes may be associated
with various human pathologies including cancer [2,3]. Even
substitutions of only one nucleotide, the so-called Single Nucleotide
Polymorphisms (SNPs) can either destroy or create microRNAbinding sites, thus, altering microRNA ability to target oncogenes
and rendering tumor-suppressor genes susceptible to microRNAmediated inhibition. By now, all human microRNA-binding sites for
all microRNAs and their target mRNAs have been predicted with
the help of the various software applications and compiled into the
databases.
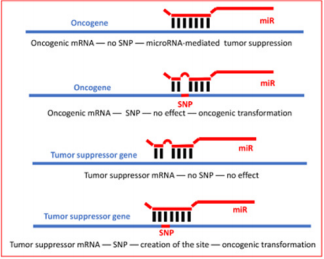
Figure 1: Destruction and creation of microRNA-binding sites by SNP; implications for oncogenesis
However, the computer-assisted prediction of microRNAbinding sites relied exclusively on one or few reference genomic sequences and did not take into consideration the significant variations within human genomes. In meantime, SNPs are reported to occur at a rate of about 1 per 300-1000 bases in the individual human genome [4] and with the frequency of 1% and higher in a population [5,6]. The number of SNP in the human genome was estimated to be around 1.5x106 per individual [7]. However, the advances of NextGen Sequencing technologies revealed that some previously believed to be rare mutations exceeded the frequency threshold set at 1% 6. Previously, we demonstrated that even the only “seed” regions of microRNA-binding sites can harbor as many as 4 SNPs, and their coincidence may result in hyper-functional or completely disabled microRNA-binding sites with the following significant phenotypic variations and predisposition to cancers [8].
Conclusion
The existing data do not take into consideration the significant
variability of microRNA-binding sites due to abundancy of SNPs. In
addition, there is no possibility to predict the creation of the new
microRNA-binding sites upon the variations of single nucleotides in
the regions, where microRNA-binding sites are absent. Finally, there
are no software tools to quickly assess the probability of
hyperfunctional or completely disabled microRNA-binding sites based on
the known frequencies of SNPs. Altogether, these problems warrant
the development of algorithms that allow biomedical researchers
to align the individual microRNA and mRNA sequences based on
their Watson-Crick complementarity in order to predict how SNPs
may weaken or enhance the known microRNA-binding sites, or to
create microRNA-binding sites de novo.
More BJSTR Articles: https://biomedres01.blogspot.com



No comments:
Post a Comment
Note: Only a member of this blog may post a comment.