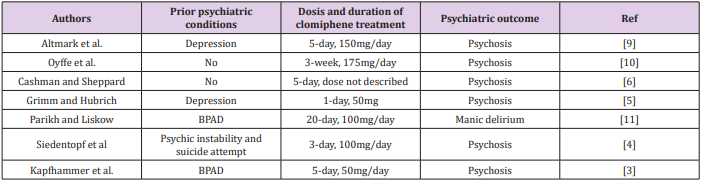
Structural and Pharmacological Properties
of Alkaloids with Special Reference to
The baine Type Alkaloids
Alkaloids
Alkaloids are pharmacologically active with two or more fused
organic compounds including heteroatoms in it. These atoms
determine the properties of alkaloids. Especially, an alkaloid
containing one or more nitrogen atoms as primary or another
form of amines may provide basicity to the alkaloid. Alkaloids
are found mainly in plants, but to the lesser extent in animals and
microorganisms [1]. Many alkaloids are toxic, but they often have
pharmacological effects. Extraction of alkaloids from plants has
been widely used by researchers for therapeutic and recreational
purposes. For example, Ephedra, opium poppies, and cocoa leaves
have been used medicinally since ancient times. In the nineteenth
century, alkaloids were first isolated successfully, and alkaloidcontaining drugs were marketed [2]. The structure of the first
alkaloid, namely coniine, was established in the year 1870 by Schiff.
Catharanthus alkaloids and paclitaxel came into the market as
researchers looked for plant drugs with anticancer properties.
Alkaloids are optically active, bitter in taste (except papaverine),
levorotatory (exception is coniine, which is dextrorotatory),
colourless (except berberine which is yellow, and harmaline and
betanidine, which are reddish), crystalline solids (except nicotine
and coniine, which are liquids), and soluble in organic solvents.
They are basic and form salts with the acids; some of the alkaloids
form as salts called quaternary amines (e.g. cinchona alkaloids with
quininic acid), while some exist as free bases (e.g. nicotine).
Alkaloids also exist as glycosides (e.g. solanum alkaloids) and esters
(e.g. atropine). The biological activity of the alkaloids frequently
depends on the amine function being transferred into a quaternary
amine at physiological pH by protonation. In summary, alkaloids
behave as curare or arrow poisons. Curare alkaloids, which have
muscle relaxant properties, are the active ingredients of arrow
poisons used by South American Indians.
Alkaloids as Medicines
In general, many alkaloids are pharmacologically active
substances which possess various physiological activities in humans
and animals. The use of alkaloids containing plants as dyes, drugs,
or poisons can be traced back almost to the beginning of civilization
[3]. Opium that derived from Papaver somniferum L. was used as
medicine in ancient Greece, Egypt and Rome. More recently, China
learned about it via Arabian traders in the eighth century A.D. Today,
a lot of alkaloids are still in use as drugs, against their poisonous
effects. Caffeine, mostly obtained from the decaffeination of coffee
species, is used a psychostimulant reagent. Codeine is used as an
antitussive. Cocaine is used as a local anesthetic, especially for eye
surgery. Morphine is an indispensable analgesic reagent that is
used for the treatment of severe pain.
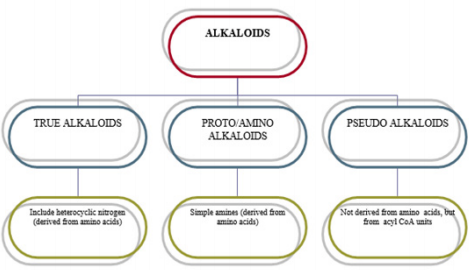
Classification of Alkaloids
Alkaloids containing wide families are classified using different
techniques [4]. These techniques: Pharmacological classification
is based on the clinical use or pharmacological activity (analgesic
and cardioactive alkaloids). Taxonomic classification is based
on the family or genus (rauwolfia and cinchona alkaloids).
Biosynthetic classification is also based on the type of precursors
or building block compounds used by plants to synthesize alkaloids
(morphine, papaverine, narcotine and colchicine may be listed as
phenylalanine- and tyrosine-derived bases). The last one, chemical
classification, is based on the chemical structure of the alkaloid.
(Thebaine, for example, is an isoquinoline derivative alkaloids of
opium; aspidospermine is an indole alkaloid.). Alkaloids are often
classified on the basis of chemical structure. These groups are
described in Figure 1.
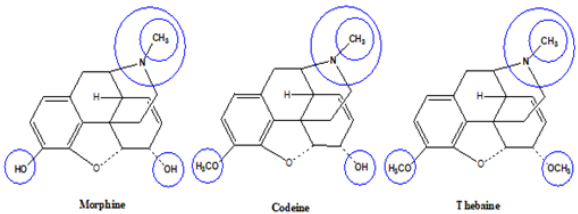
Opioid Structure
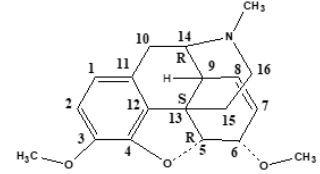
Morphine includes a benzene ring with a phenolic hydroxyl
group at 3-position, an alcohol hydroxyl group at 6-position, and
the nitrogen atom in Figure 2. When both hydroxyl groups are
converted to ethers, codeine and thebaine as morphine derivatives
are obtained. The tertiary form of the nitrogen appears to be crucial
to the analgesic properties of morphine. The nitrogen quaternary
widely decreases the analgesia, since morphine cannot pass
into the central nervous system. Also, if the methyl group on the
nitrogen is changed, it will again decrease analgesia. Morphine is
also optically active, and only the levorotatory isomer exhibits the
analgesic effect. Furthermore, they have many effects on the central
nervous system. These are analgesia, respiratory depression,
euphoria, and depression of cough reflex. Morphine undergoes
strong first-pass metabolism by the liver if taken orally, limiting
the effective time of analgesia, unless a sustained-release form is
given [5]. The adverse effects of morphine include CNS respiration
depression, sedation, dizziness, nausea, vomiting, itching, and
constipation. CNS respiratory depression is the serious one among
other complications.
HRM Data Analysis
HRM data analysis was performed using the LightCycler® 96
SW 1.1 software (Roche Diagnostics, Germany). All analyses were
carried out including both positive (on the second and the third
plates/SSR marker) and negative controls. The positive controls
represented the different groups detected mostly on the first, then
the second and then the third plates, and finally on a forth plate in
case a new different group was discovered at the third plate. They
were used to enable grouping the accessions among the three runs
of each marker because the software does not support analyzing all
data gathered from these runs together. After normalization, samples
having the same melting curves were grouped automatically, with
sensitivity settings at the default level 50%. However, grouping was
also evaluated by visual inspection of the HRM profiles produced
and corrected manually if needed through the adjustment of the pre
and post-melting ranges by dragging the two leftmost and the two
rightmost vertical sliders to appropriate locations. Generally, they
were adjusted as close as possible to the melting temperature.
Apart from these severe effects, morphine is still considered to
be the most effective drug clinically available for the management of
severe pain associated with cancer. The opium poppy was cultivated
in Mesopotamia around 3400 BC. Opium is a mixture of alkaloids
from the poppy seed. Opiates are naturally occurring alkaloids
such as morphine or codeine. An opioid is a term used broadly to
describe all compounds that work at the opioid receptors [6]. The
common term narcotic (from the Greek word for stupor) originally
was used to describe medications for sleep, and then was used to
describe opioids, but now is a legal term for drugs that are abused.
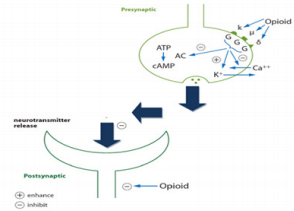
Opioid Receptors
Definition of an Opioid Receptor
Thus, the opioid is a substance that produces the above-listed
effects by acting at opioid receptors and whose actions are reversed
by naloxone.
a) Extraction/purification of active poppy ingredient: morphine
[7].
b) Effects: analgesia, respiratory depression, somnolence,
decreased GI motility.
c) Agonist structure-activity relationships: morphine ≥
meperidine > codeine > 0
d) Structurally similar agents such as naloxone act as antagonists;
have no effect alone but reverse the effects of an agonist.
e) Opioid binding interactions showed high-affinity binding sites.
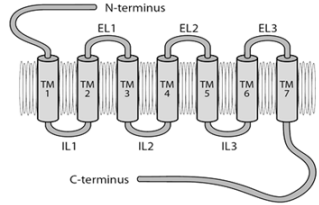
Opioid Receptor Subtypes
Three types of opioid receptors have been identified so far: mu
(µ) receptors, delta (∂) receptors, and kappa (κ) receptors (Figures
3 & 4). Mu (μ) (agonist morphine) Mu receptors are found mainly in
the nervous system (brainstem and medial thalamus). Mu receptors
are responsible for respiratory depression, supraspinal analgesia,
sedation, euphoria, sedation, decreased gastrointestinal motility,
and physical dependence. Subtypes of Mu receptors possess Mu1
and Mu2. Mu1 is confined to analgesia, euphoria, and serenity, while
Mu2 is concerned with respiratory depression, prolactin release,
pruritus, dependence, anorexia, and sedation. These receptors are
called OP3 or MOR (morphine opioid receptors). Kappa (κ) (agonist
ketocyclazocine) Kappa receptors are found in the limbic and
other diencephalic areas, brain stem, and spinal cord, and induce
spinal analgesia, sedation, dyspnea, dependence, dysphoria, and
respiratory depression. These are also known as OP2 or KOR (kapa
opioid receptors). Delta (δ) (agonist delta-alanine-delta-leucineenkephalin) Delta receptors are noticed in the brain, and their
effects are not clearly explored.
They may be responsible for psychomimetic and dysphoric
effects. They are also called OP1 and DOR (delta opioid receptors).
Sigma (σ) (agonist N-allyl nor metazocine) Sigma receptors are
associated with psychomimetic effects, dysphoria, and stressinduced depression. They are not included as opioid receptors, but
rather are the target sites for phencyclidine (PCP) and its analogs.
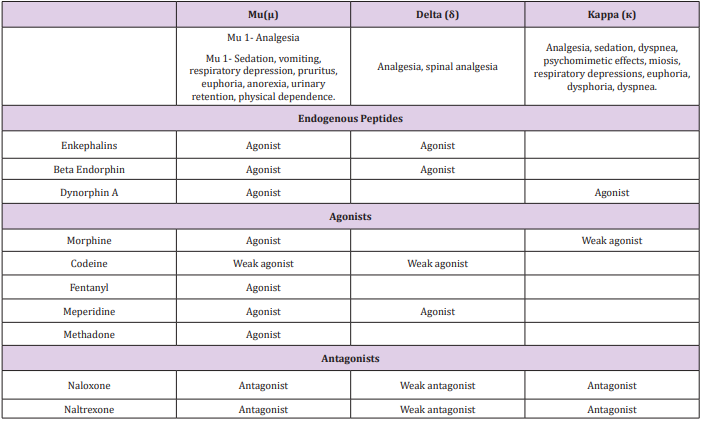
These receptor types are defined by:
1) Agonist structure-activity relationships in the bioassay (ability
to block stimulated contraction of specific smooth muscle
tissues) and binding (Table 1).
2) Antagonist activity profile: naloxone blocks all opioid receptors
(Table 1).
3) Lack of cross-tolerance.
4) Cloning
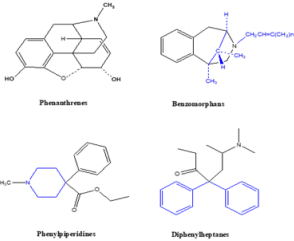
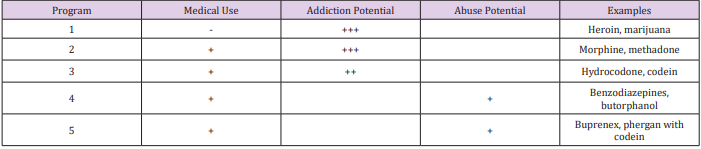
Opioid Classes
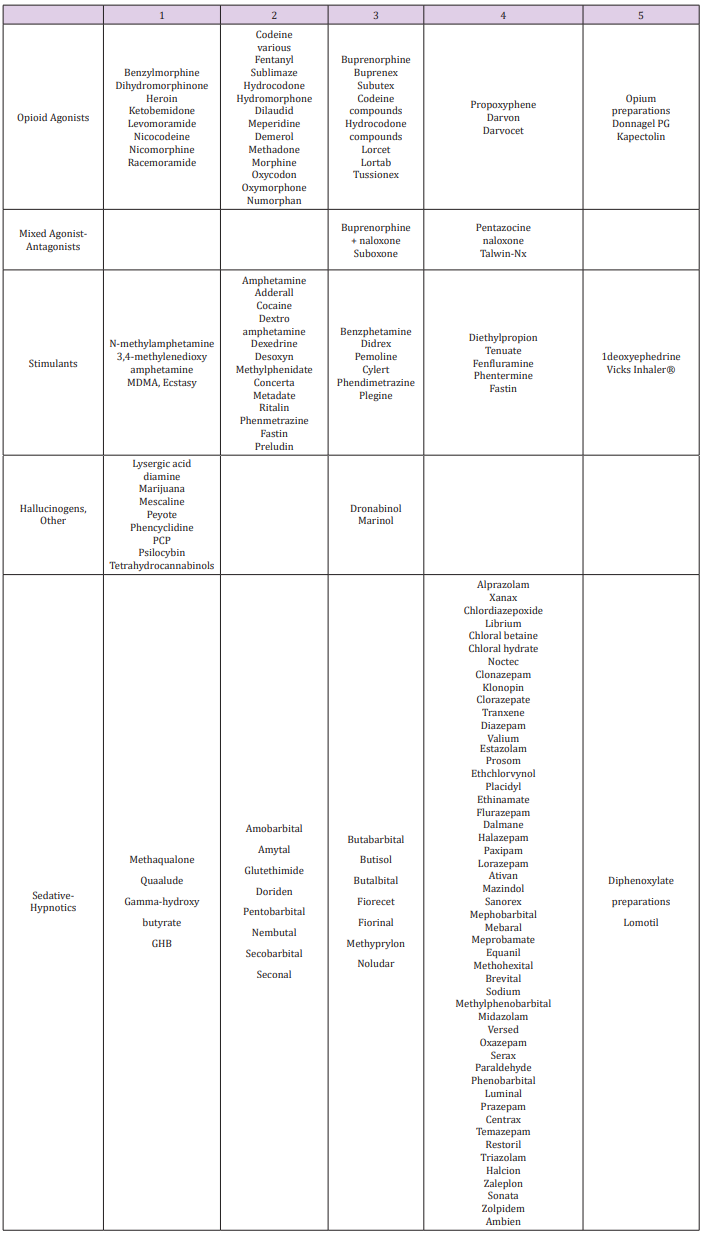
Opioid Classes DEA (Drug Enforcement Agency) classified
opioids into schedules as illustrated in Table 2 and Table 3. Four
chemical classes of opioids (Figure 5): Phenanthrenes are the
prototypical opioids. The presence of a 6-hydroxyl is associated
with nausea and hallucinations. Morphine and codeine (both
with 6-hydroxyl groups) are associated with more nausea
than hydromorphone and oxycodone (which do not possess
6-hydroxyl groups). Opioids in this group include morphine,
codeine, hydromorphone, levorphanol, oxycodone, hydrocodone,
oxymorphone, buprenorphine, nalbuphine, and butorphanol.
Benzomorphans have pentazocine as a member of this class and
is an agonist/antagonist with a high incidence of dysphoria.
Phenylpiperidines cover fentanyl, alfentanil, sufentanil, and
meperidine. Fentanyl has a strong affinity for the mu receptor.
Diphenylheptanes include methadone and propoxyphene.
Tramadol does not come under standard opioid classes. Tramadol
is an atypical opioid, a 4-phenyl-piperidine analogue of codeine,
with partial mu-agonist activity, in addition to central GABA,
catecholamine, and serotonergic activities.
Opioids can be classified agonist, agonist-antagonist, partial
agonist, or partial antagonist. Compounds can have intrinsic affinity
and efficacy at receptors, with affinity being a measure of the
strength of interaction between a compound and its receptor and
efficacy being a measure of the strength of activity. An agonist has
both affinity and efficacy; an antagonist has affinity but no efficacy;
a partial agonist has affinity but only partial efficacy. Regarding
the opioids, the relevant receptors are the mu, kappa, and delta
receptors. Compounds can have differing degrees of affinity and
efficacy at these various receptors.
Medicinal Uses and Toxicity of Thebaine and its
Derivatives
Alkaloids protect plants against bacteria, fungi, insects, and
herbivores, as well as other plants by means of allelopathically
active chemicals. Thebaine, or paramorphine, is a white, crystalline,
slightly water-soluble, poisonous alkaloid. This alkaloid type is not
used for therapeutic purposes but is converted to other compounds,
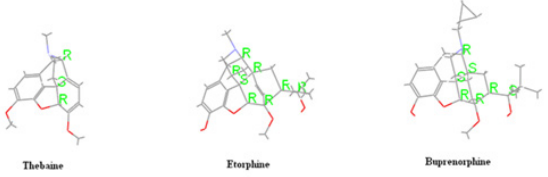
such as buprenorphine, etorphine, oxycodone, oxymorphone, and
naloxone. Thebaine derivatives are used for different purposes.
5[alpha])-6,7,8,14-tetradehydro-4,5-epoxy-3,6-dimethoxy-17-
methylmorphinan is more toxic and is about ten times higher than
morphine. Also, it is the most poisonous opium alkaloid. Because
of this reason, thebaine is used only for synthesis into other
pharmaceutical drugs. Usually, thebaine readily undergoes Diels–
Alder reactions with various dienophiles to form into adducts.
The diene system of thebaine could potentially be attacked from
both sides, but reactions with dienophiles always occur from the
same face as the nitrogen bridge (upper face) due to the nitrogen
bridge causing the lower face to be hindered through concealment
inside a concave system [9]. The nature of a substituent in
positions 7,8 of morphine alkaloids is among the most important
factor affecting their biological activity [10]. The opioid analgesic
buprenorphine, ethorphine possesses a pharmacological profile
that may be interesting for development of antinarcotics [11]. Our
previous reports show that cinnamaldehyde, a plant derivative,
is an allergen, and it induces toxicity [12,13]. In this regard, an
attempt has been carried out to evaluate the toxicity of thebaine,
an allergen [14].
Hashish capsule (Papaver semniferum L.) contains nearly
thirty kinds of alkaloids. Among this morphine, thebaine, papaverin
and codeine are important to medicine raw materials that have
medicinal value. In pharmacy, thebaine cannot be used directly
as a drug because it exhibits toxicity but thebaine has a very
important role as the main material with some reasons [15,16].
These reasons include its view availability, its lower cost and
chemical structure, containing a conjugated diene system at ring C.
The chemical structure of thebaine has allowed the preparation of
many pharmaceutical products by Diels-Alder cycloadditions with
a large number of dienophiles. Further, classical examples of drugs
prepared using this approach are Etorphine [17] (Immobilon),
Buprenorphine [18] (temgesic, buprenex, Buprex, Prefin) and
many other adducts, reported mainly by Bentley [19-21] (Figure
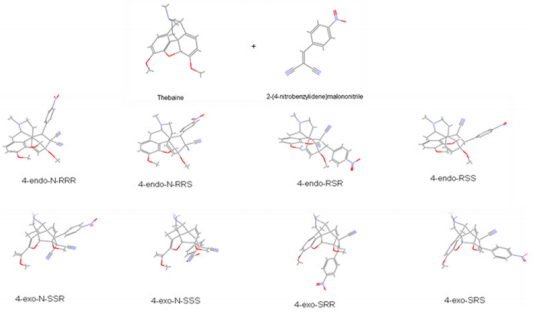
6). Surprisingly, no references are found in the literature on
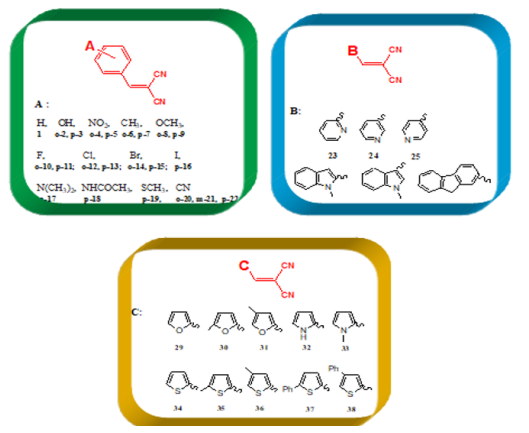
the cycloaddition to thebaine of the dienophile that are various
benzylidene malononitrile (1-28) and metilen malononitrile (29-
38), (Table 3).
Therefore, we firstly performed PM3 and DFT methods to
evaluate the best dienophiles by using Gaussian09 [22] and
Discovery Studio 3.5, (DS 3.5) [23]. In the meantime, we examined
the effect of dienophile substitution (38 molecules) on the reactivity
to select the substituents leading to the most favourable suitable
reaction conditions. Then, novel and efficient addition products
(Table 4) were analysed by using quantum chemical descriptors
[24] for investigation of the differences in stereochemistry and
determination of the energetically more suitable reactions.
The quantum chemical descriptors have proven useful for the
prediction of many molecular biological and physicochemical
properties of interest to the pharmaceutical industry [25-27]. In
conclusion, the obtained results about the known basic structure,
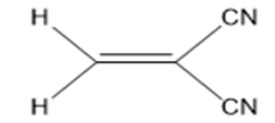
2-methylenemalononitrile, (MMN) (Figure 7), and the 38 molecules
(Table 4) are evaluated to obtain a better understanding of the
relationship between structure and activity. The same process is
also applied to the novel addition products (Table 5), basing on
thebaine and known drugs such as etorphine [17], buprenorphine
[18], (Figure 6).
Thebaine has similar anesthetic activity as morphine, but it
is very toxic and is obtained very little, compared to morphine.
These reasons are generated from different searches on chemical
modification of thebaine to eliminate and diminish side effects
and also to increase the activity. Firstly, the various benzylidene
malononitrile (1-28) and metilen malononitrile (29-38), (Table 4)
as dienophiles, and possible semi-synthetic thebaine derivatives,
(38*8 = 304 compounds) (Table 4) were designed and optimized
with semi-empirical PM3 and B3LYP/6-31G* level of theory in DFT
methods by using Gaussian09 [22] and DS 3.5 [23] software. All
stationary points were located and characterized as true minima
by using both package programs with the semi-empirical PM3
and B3LYP/6-31G* basis sets. The calculated quantum chemical
descriptors were total energy (E), the energy of the highest occupied
molecular orbital (HOMO), the energy of the lowest unoccupied
molecular orbital (LUMO), the global hardness (η), electrophilicity
index (ω), electronic charge (ΔN) and dipole moments (DM).
Tables 1 & 2 for various dienophiles and Tables 3 & 4 for
novel addition products were shown in Appendix. Furthermore,
the calculated results were divided into two parts such as the
dienophiles (Table 4) and novel addition products (Table 5). Each
part was explained with figs (Figures 2-9). In the first part, the
dienophiles were clarified based on 2-methylenemalononitrile
(MMN) (Figure 7). In the second part, new thebaine derivatives were
interpreted according to the obtained results with comparison to
thebaine and known drugs such as etorphine [17], buprenorphine
[18] compounds (Figure 6).
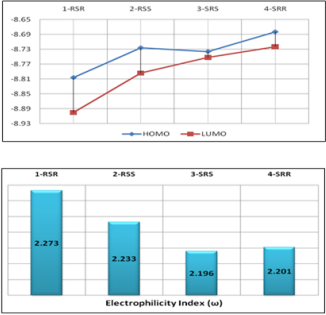
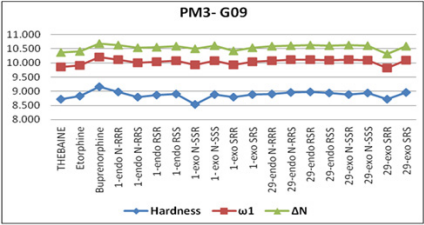
In the literature, the activity value and active configuration of
thebaine were 2.51 and RSR form (Figure 8). Because of this, all
possible configurations of thebaine were drawn and optimized the
PM3 level of a semi-empirical method by using DS 3.5 [23] and
Gaussian09 [22]. It is indicated that RSR form of thebaine was
more stable than others with the help of various quantum chemical
descriptors such as HOMO, LUMO and electrophilicity index,(ω),
(Figure 1). Also, the difference between HOMO and LUMO of RSR
configuration of thebaine has rather bigger magnitudes. The
calculated electrophilicity index value of thebaine is also 2.273. As
a result, all descriptors are validated and represent the stability of
RSR form of thebaine in all possible configurations of thebaine. In
the other words, theoretical calculations favor the experimental
results of thebaine in literature (Figure 9).
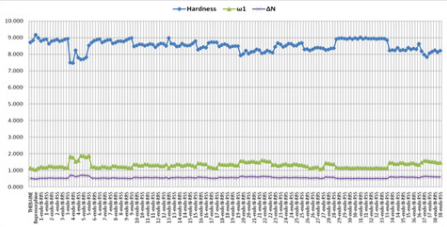
The various dienophiles (1-38):
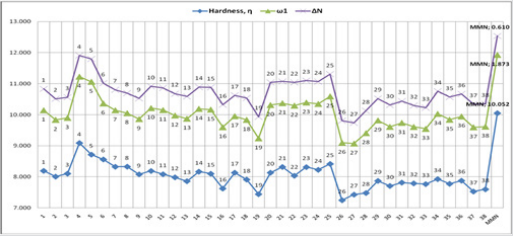
According to the obtained data from Gaussian09 and DS 3.5
software, the global hardness (η), electrophilicity index (ω), and
electronic charge (ΔN) values were calculated for the dienophiles
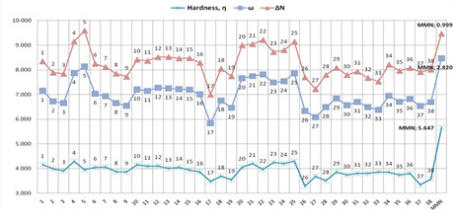
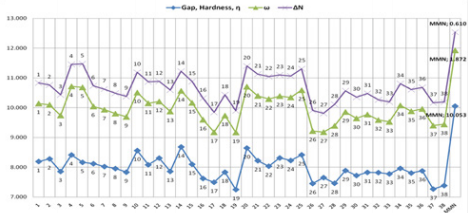
(Table 3), and MMN (Figure 7). These results are listed in Figures
10-13. Within Table 1, there are three frames including six or
fivemembered aromatic heterocycles monosubstituted by electronacceptor
and –donor groups. These frames are entitled A, B and
C. The structural as well as electronic characteristics induced by
chemical substitution are because of the different responses of the
global electrophilicity power. For instance, 2-(4-nitrobenzylidene)
malononitrile, (p-5) has a prominent difference for all quantum
chemical descriptors in all dienophiles (1-38) and MMN. Because
nitro is a strong electron-withdrawing group, it increases the
electrophilic character. The -NO2 substituted benzylidene
malononitrile structures, (o-4; p-5) have also shown the highest
tendency in the electrophilicity index (ω) and electronic charge
(ΔN) values. The second higher electrophilicity index (ω) value
belongs to –CN-substituted structures (p-22).
Meanwhile, other descriptors favor this opinion, (Figures 10
& 11). On the contrary, the global hardness (η) value shows the
lowest trend on A, B, and C frame shapes. In the first structure A,
for 2-benzylidenemalononitrile (1), has higher electrophilicity
index (ω) and electronic charge (ΔN) values, [ ω: 1,949; ΔN: 0,690]
compared to MMN as standard reagent [ ω: 1,873; ΔN: 0,610]. For
2-benzylidenemalonitrile (1), the substitution of a hydrogen atom
by a strong electro-donor group, such as -OH, (o-2; p-3) reduces the
electrophilic character. Likewise, upon substitution of the hydrogen
atom by different electro-donor groups (-CH3 (o-6; p-7) and -OCH3
(o-8; p-9)), the electrophilicity power further decreases. The –I
(p-16), –Cl (o-12; p-13), –Br (o-14), –F (o-10), –Br (p-15), and –F
(p-11) substituents classified in increasing order of electrophilicity
power (ω), (Figure 10). As known, when the chemical substitution
in the dienophile groups is increased by electro-donor groups,
global electrophilic character decreases.
For example, (E)-2-methyl-3-(1-methyl-1H-indol-3- yl)
acrylonitrile (p-27) has the worst behaviour of all quantum
chemical descriptors in all dienophiles (1-38) and MMN. On the
other hand, when electro-withdrawing groups (-SCH3, -N(CH3)2,- NHCOCH3, and -CN)s’ electrophilicity power further increases,
it indicates an increase of reactivity towards a nucleophile. In
addition to this, para position in any structure is preferred rather
than ortho and meta positions for all dienophiles (1-38). As with
the benzylidene analogues, replacement of pyridine (23, 24, 25) ,
indole (26; 27) and carbazole (28) in second group B, compound
25, which contains nitrogen group is also placed in a para position,
exhibits good activity in others, (Figure 10), (see detail information
Table 1 in Appendix.). In the last group C, including five-membered
aromatic heterocycles monosubstituted by electron-acceptor and
–donor groups, (Figure 10) demonstrates the decreasing order
of electrophilicity index (ω) and electronic charge (ΔN) values,
(thiophene > furan > pyrrole), respectively
For furan and thiophene compounds, the substitution of a
hydrogen atom by an electro-donor group, –CH3, gives increase
when –CH3 places to meta position instead of ortho position.
However, upon substitution of a hydrogen atom on nitrogen
by –CH3, decreases the activity based on the pyrrole. If –CH3 is
replaced with –Ph group, these structures (37, 38) have the higher
increase in group C. In addition, it mentioned nearly the same trend
for the dienophiles and MMN at B3LYP/6- 31G* level of theory in
DFT method by using Gaussian 09 software (Figure 11). At the
same time, the global hardness (η), electrophilicity index (ω), and
electronic charge (ΔN) values were calculated for the dienophiles
and MMN by using DS 3.5 software (Figures 12-13). As a result, the
same behaviours of investigated compounds are shown (Figure 10
vs. Figure 12). Also, these results are rational and coherent for the
various dienophiles and MMN, according to different methods such
as PM3 and B3LYP/6-31G* level of theory in DFT methods by using
Gaussian 09 software and PM3/VAMP and BLYP/DMol3 methods
by using DS 3.5 software.
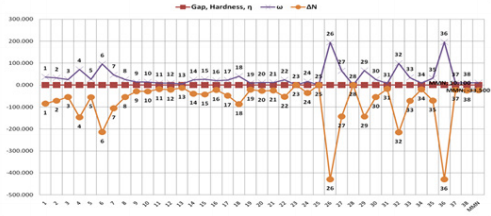
In particular, the obtained results with PM3 and PM3/VAMP
methods have similar numerical values and trends (Figures 10 &
12). However, there are logical and possible results with B3LYP/6-
31G* basis set (Figure 11). The results with BLYP/DMol3 method
by using DS 3.5 software shows inconsistent and lacking correct
logical relation data for the dienophiles and MMN (Figure 13).
Furthermore, PM3 method in Gaussian 09 program is implemented
for additional novel products in the further process of our work,
because PM3 and PM3/VAMP methods in Gaussian 09 and DS 3.5
software have similar behaviour. However, BLYP/DMol3 method
by using DS 3.5 software also exhibits extraordinary results. In
Gaussian 09 software, PM3 and B3LYP/6-31G* levels of theory in DFT methods show consistency in their results, but the additional
novel products are not suitable for analysis at B3LYP/6-31G* level
of theory in DFT method by using Gaussian 09 program, due to
rather huge and steric groups of studied compounds, (molecular
weight: 400 – 600 g/mol).
Novel Addition Products
In the second part of the study, we first investigate and
understand the differences in stereochemistry and predict the
reactivity of thebaine and its novel and efficient addition products
(Table 4) by using quantum chemical descriptors [24]. The new
thebaine derivatives were evaluated according to the obtained
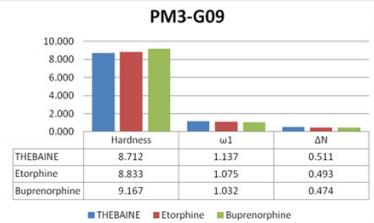
results with comparison to thebaine and known drugs, (Figure
6). The calculated descriptors of thebaine and known drugs were
also given in Figure 14. Moreover, the diene system of thebaine
could potentially be attacked from both faces, yet reactions with
dienophiles occur from the same face as the nitrogen bridge
(upper face) due to the nitrogen bridge causing the lower face to
be hindered through concealment inside a concave system. Thus,
all possible configurations of additional novel products were
computed at PM3 method by using Gaussian 09 software (Figure
15). This method was selected based on its attainable, suitable, and
rational results in the former calculations for dienophiles (Table
3). It is known that global hardness provides information about
stabilities of compounds [24].
Electrophilicity index provides comprehensive information
about the structure, properties, stability, reactivity, interactions,
bonding, toxicity, and dynamics of many-electron systems
in ground and excited electronic states [27]. With respect to
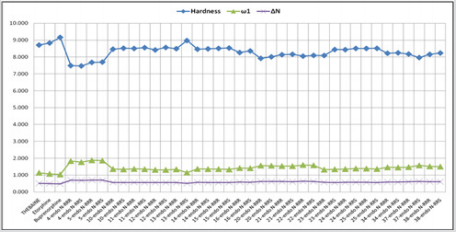
the obtained results, Figure 16 exhibited the trend of three
quantum descriptors values of possible eight configurations of
2-benzylidenemalononitrile (1) in 6-ring compounds and 2-furan2-ly-methyl malononitrile (29) in 5-ring compounds in comparison
to thebaine and known drugs. (See detail information Tables 3 & 4 for
additional novel products in Appendix.) It showed that endoforms
of 6- and 5-ring compounds were more stable than exoforms and
more effective than thebaine and known drugs. As expressed above,
endo forms of novel addition compounds were more stable than
exo-form; thus endo forms of the compounds were separated in
data. Then, the compounds include electron-withdrawing groups,
exhibited better performance than the compounds that include electron-donating groups, such as -NO2 versus –OH. Afterwards, all
investigations were applied only to the selected compounds.
These were 4, 5, 10-16, 20-25, 34, 37, 38, given as Table 5 in
Appendix and the obtained results were represented in Figure
17. The Figure 17 shows that endo forms of 4 and 5 compounds
have predominant properties compared to others in all quantum
chemical descriptors. Also, the selected compounds were more
effective than thebaine and known drugs (Figure 6).
In summary, the global electrophilicity index introduced by
Parr et al. [28-30] becomes a useful means of understanding
the substituents in the diene/dienophile interacting pair. It also
provides a way to classify the electrophilicity power of a series
of dienophiles, the addition novel products of thebaine. Upon
increasing dienophile substitution with electro-withdrawing
groups, –F, –Cl, -Br, -I, (10-16), –CN, (20-22), and –NO2 (4-5)
substituents, there is an increase in ω index, thus reflecting a higher
electrophilic character. The structural and electronic characteristics
induced by chemical substitution are due to different responses of
the global electrophilicity power. The influence of the combination
as well as the positional relationship of the substituents of each
dienophile and the addition novel products can be analysed to
develop most suitable reactions for the design of possible precursor
to be integrated into synthetic sequences. Results are in agreement
with those experimentally developed for each diene/dienophile
pair in D-A reactions. Depending on the observed global hardness
(η), electrophilicity index (ω), and electronic charge (ΔN) values,
it can be assumed that for various benzylidene malononitrile and
metilen malononitrile, the substitution of a hydrogen atom by a
strong electro-withdrawing group, such as nitro –NO2 (4-o and
5-p), gives rise to an increase of the electrophilic character.
The authors acknowledge Y. Yildirir and F. Sevin Duz for data
collection and E. Aki-Yalcin and ESIS research group for technical
support.
For more Articles: https://biomedres01.blogspot.com/