Breast Cancer Prognosis: A Genetic Code for Personalized Therapy
Introduction
Today, breast cancer is distinguished into at least three different
subtypes based on clinical and molecular parameters: luminal,
erbB2, and basal type, which exhibit different biological behaviors
and prognoses. Correctly identifying the molecular subtype of
the tumor opens the door to new, increasingly adequate and
targeted therapeutic possibilities for the treatment of the specific
molecular subtype [1]. In this scenario, luminal carcinomas (which
represent approximately 65% of the total) are distinguished by a particular heterogeneity of biological behavior, with recovery
of disease in approximately 40-50% and death in approximately
two-thirds of these patients 5 years after diagnosis, despite initial
anatomopathological pictures of apparent low aggressiveness [2-
9]. Precisely for this diagnostic category, biomolecular parameters
derived from the genome/transcriptome that are capable of
orienting the therapeutic choices in a more precise and personalized
way on the patient’s actual therapeutic needs are desirable
[10,11]. Currently, the clinical (T, nodal status) and biopathological
(hormonal status) parameters obtained from membrane receptor
expression provide prognostic information and an indication of any
adjuvant systemic chemoradiotherapy treatment [12-14]. However,
adjuvant therapy reduces the risk of recurrence by only 25-30%
[13]. These data are probably due to:
1. Clinicopathological parameters of stratification of the risk of
disease recovery are not sufficiently adequate for the patient’s
prognostic framework.
2. Adjuvant therapies are not specific enough toward the cells
responsible for disease recovery [15,16].
Knowing the details of the mutations of every single tumor
allows us to predict the biological behavior of that neoplasm and
to adequately stratify the risk. Genetic tests, such as Mammaprint
and Oncotype DX, EndoPredict [17,18], assist clinicians in
choosing the most suitable adjuvant treatment by analyzing the
expression profile of genes involved in the metastasis process (St
Gallen 2017). These tests are very expensive and often have to be
sent to foreign laboratories. The genetic profile is of the utmost
importance in the evaluation of parameters already known as the
expression of hormonal receptors and HER2; these are currently
determined with immunohistochemistry (IHC) or FISH methods,
which provide information about the morphological expression
of the receptor but not about their functional state. In any case,
knowing that the receptor is expressed at the membrane level is
not sufficient information to guarantee the effectiveness of the drug
addressed to it because that protein may not be functionally active.
Therefore, it is necessary to ascertain the functional activation of
the gene responsible for the synthesis of the protein to guarantee
its functionality, more than its presence. Moreover, from the
gene expression profile, additional information that specifically
correlates the expression of some genes to the response to
individual therapies can be obtained; for example, in HER2-positive
patients, the high expression of IGF1R correlates with resistance to
Herceptin, as well as the hyperexpression of CCNE1; instead, ER+
patients with high PDGFRA expression are resistant to tamoxifen
treatment [14,19,20]. The use of gene profiling tests offers the
opportunity for a more adequate risk stratification, an improvement
of the therapeutic planning and the clinical outcome, avoiding what
happens today, with the standard clinical-pathological criteria,
namely, the undertreatment of approximately 20% of women with
grade 1 breast cancer and overtreatment of approximately 15% of
women with grade 3 breast cancer
Methods
This retrospective study aimed to observe the difference in the receptor state obtained from the genome compared to that obtained with traditional immunohistochemistry. Analyzing biological material from formalin samples from 1994/1995 from two groups of selected patients with breast cancer, one consisting of patients still alive and the other of patients with a survival of less than 4 years. Survival data of a group of patients with a long follow-up period (RTUP) suffering from breast cancer diagnosed in the twoyear period 1994/1995 were obtained with the authorization of the Ethics Committee of Umbria (CEAS). The group, consisting of 55 patients, was then divided into 2 numerically balanced subgroups: the first of 30 patients staged alive on 31/12/2013 with survival of >20 years and the second of 25 patients staged in death with lower survival 4 years after diagnosis (Table 1). The relative tissue samples of the respective patients fixed in formalin and included in paraffin (FFPE) were subsequently collected and stored in the histoteca of the SC of Anatomy and Pathological Histology of the Hospital of Perugia. Twenty-two patients who had the respective tissue sample unsuitable for lack of biological material in the original inclusion block were excluded. The remaining samples, for a total of 33 cases, were all characterized again from a biopathological point of view through a dedicated immunohistochemical panel: ER, PgR, Ki67, HER2 and subjected to microscopic evaluation by a pathologist who was thus able to identify two groups of carcinomas: luminal and nonlluminal (according to the San Gallen criteria 2011 and following).
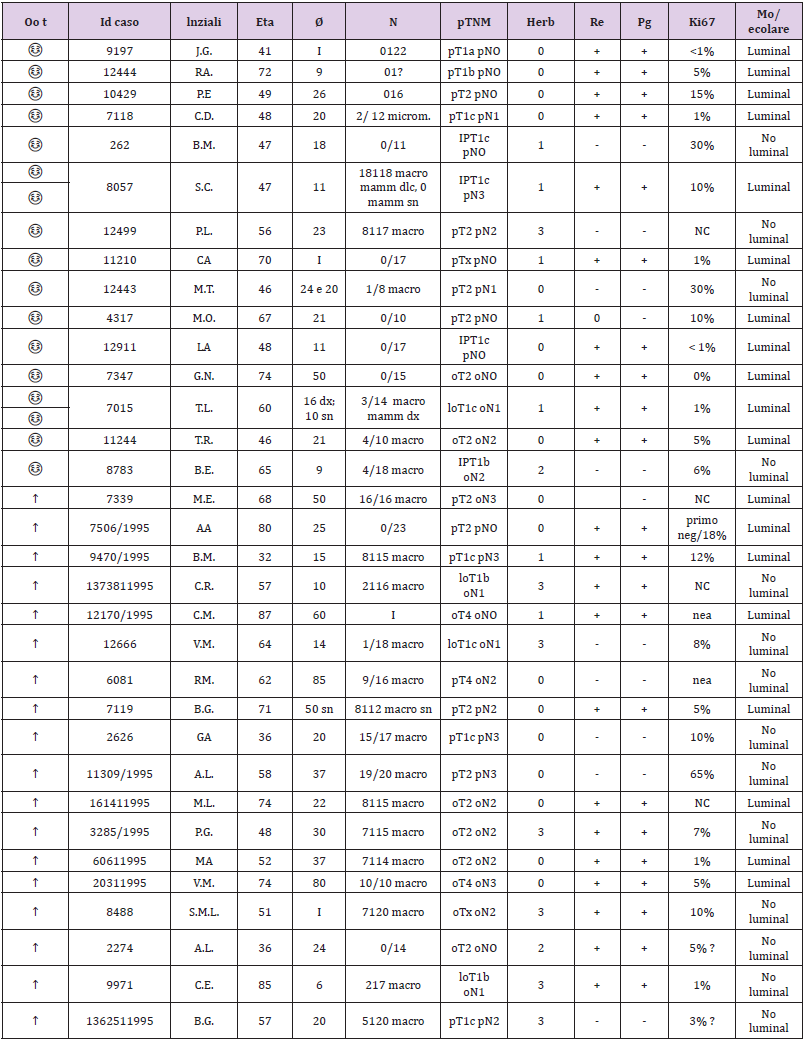
Table 1: The two-subgroup division of the 55 patients.
The recharacterization provided for the preparation of 1 slide
for hematoxylin and 4 slides with blank sections necessary for the
immunohistochemical panel for each sample. The pathologist made
the diagnosis based on current guidelines and verified that in each
section stained with the hematoxylin of each patient there were at
least 30% of the neoplastic cells out of the total. This percentage
figure is necessary to maximize the extractive yield of total RNA.
In practice, for samples with a number of neoplastic cells ≥ 30%
of the total, it was not necessary to proceed with macrodissection.
In the first phase of the experimentation, so with the 33 starting
samples, the enrichment of the sample was not necessary, but 2/4
sections of the FFPE fabric of 10 μm thickness were cut in sequence
on microtome and placed in 1.5 ml safe look tubes ready to be
extracted. Total RNA, after appropriate quantitative and qualitative
evaluation, was used for the preparation of cDNA for sequencing
purposes. The library used is Illumina’s TruSeq Total RNA. In the
second phase of the experimentation, 2 pilot samples were used to
validate the panel of genes obtained with the first phase; they were
homogeneous both as biopathological characteristics of the lesion
(early breast cancer; tumor diameter; lymph node status; state of
HER2 and so on) and as the age of the patients. In the end, for each
patient, we obtained 2 tubes of tumor tissue and 2 tubes of healthy
tissue ready to be extracted. The extraction kit used to obtain total
RNA was the same as that used in phase I of the trial. Total RNA,
after appropriate quantitative and qualitative evaluation, was used
for the preparation of cDNA for sequencing purposes. The library
used was Illumina’s TruSeq RNA Access, suitable for processing
RNA extracted from paraffin samples. Total RNA was extracted
from sections of paraffin tissue using the “Tissue Preparation
Reagents” kit - Sividon Diagnostic, from the Pathological Anatomy
Section of the Santa Maria Della Misericordia Hospital. The chosen
extraction method was previously “validated” on 2 samples with
characteristics of “age of the sample”, “type of tissue” and “origin”
identical to the samples to be used for this work. Validation test of
the extraction method suitable for the preparation of the “TruSeq
RNA Sample Prep” library from RNA for sequencing.
For the deconvolution of the data from the HiSeq sequencer, to
process them, the following steps were carried out:
a. Demultiplexing: phase necessary to attribute to every single
sample its respective data (1 sample = 1 fastq file).
b. Fastqc: phase in which the quality control of the sequencing is
carried out through the use of the “fastq file for reads quality”
tool.
c. Trimming: delicate but essential phase of the deconvolution
process because it eliminates the reads and low-quality
fragments from the Fast file.
d. Mapping: important phase of the process because for each
sample a same file (also called bam) is built which specifies
how the reads align on the reference genome.
e. Count table assembly: a final phase that includes all the
information from the same file of the analyzed samples and
related “count reads” in a single table.
Based on the reference human genome, the following are the
data of the number of reads that align: Human Genome Assembly
(GRCh37/hg19). These data are used as a criterion for deciding
which samples will be analysed.
Samples with a low quantity of exons were excluded: a low
number of reads mapped to exonic regions <12%.
Two R/Bioconductor 3.2.2 packages were used for statistical
analysis:
1. DESEq2 (Love MI, Huber W, Anders S. Moderated estimation
of fold change and dispersion for RNA-seq data with DESeq2.
Genome Biol. 2014; 15 (12): 550.)
2. edgeR (Robinson MD, McCarthy DJ, and Smyth GK (2010).
“edgeR: a Bioconductor package for differential expression
analysis of digital gene expression data.” Bioinformatics, 26,
pp. -1)
The edgeR package was used on the data analyzed with DESeq2
to confirm the results obtained with this calculation algorithm. Both
are based on the negative binomial distribution but use different
correction tests. The use of the two packages allows obtaining a
list of genes that for both calculation algorithms are differentially
expressed between the samples.
The statistical significance that will be used in the two packages
is shown below:
1. DESeq2: P-value <0.05; P adjusted value <0.1
2. edgeR: P-value <0.05; False Discovery Rate <0.1.
An analysis of the network of genes significantly differentially
expressed in the two groups was also conducted using GeneMania
(http://www.genemania.org). This network was built based on the
interactions between the genes, as reported by results published
in the literature. Features such as gene coexpression, proteinprotein
interactions, physical interactions between genes and
other functions as described in the studies indicated in the table
compared to the network figure are evaluated. Although paraffin
introduced non negligible “background noise” in the analysis, the
number of reads obtained was satisfactory to conduct a differential
expression analysis using a “Read Counter” approach. It is possible
to evaluate the formation of 2 small subclusters, and the analysis of
differential expression with this dataset allows us to differentiate
25 genes differentially expressed between the 2 groups. The
hierarchical cluster of significant genes shows a homogeneous
trend among the 2 groups except for 2 luminal samples, which,
although clustered with nonluminal samples, are very close to
samples of the same stage (sample 7506 luminal staged due to
death is very close to sample 2626 nonluminal staged due to
death; sample 10429 luminal staged in life is close to sample 262
nonluminal staged in life). The IFIT3 and MX1 genes, among the
25 differentially expressed genes, have a strong interactome with
other genes not present in the list but strongly correlated with
each other in biological processes.From a careful observation of the PCA
luminal vs nonluminal of the samples selected in Figure 1, it is
possible to observe the formation of 2 groups of samples that do
not comply with the luminal/nonluminal classification provided,
as shown by the PCA. Although paraffin introduced nonnegligible
“background noise” in the analysis, the number of reads obtained
was satisfactory to conduct a differential expression analysis
using a “Read Counter” approach. It is possible to evaluate the
formation of 2 small subclusters, and the analysis of differential
expression with this dataset allows us to differentiate 25 genes
differentially expressed between the 2 groups. The hierarchical
cluster of significant genes showed a homogeneous trend among
the 2 groups. Among the genes, the FGFR3 gene is highlighted: a
2012 paper may be useful in which the following is stated: “FGFR3
activation in MCF7 cells stimulated activation of the mitogenactivated
protein kinase (MAPK) and phosphoinositide 3-kinase
(PI3K) signaling pathways, both of which have been implicated in
tamoxifen resistance in breast cancer”. It should be considered that
it is 4 times less expressed in “luminal” cases than in “nonluminal”
cases.
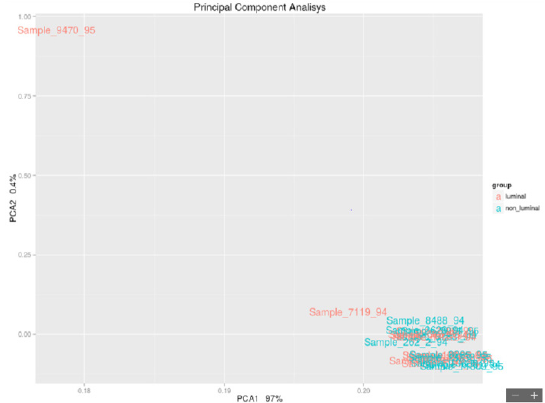
Figure 1: Luminal vs Non-Luminal.
Results
From the comparison of the results obtained with the two
methods of statistical analysis, 62 significant genes emerged
between living and deceased patients (Figure 2). In particular, 12
overexpressed genes emerged in live patients, and 50 overexpressed
genes emerged in deceased patients. The 8 intersecting genes of the
diagram are IFITM10, CEACAM7, KCNE4, OGDHL, NPY1R, SNCG,
ARHGAP23, and PIEZO2. The unique DESeq 2 gene is RBFOX1.
In this study, the data of the 26 samples (previous analysis)
were analyzed again to highlight the genes that are differentially
expressed in the luminal (life/death) and in the nonlluminal (life/
death) samples using the bioinformatic tools edgerR and DESeq2
in the R/Bioconductor environment using the following settings:
DESeq2 pValue <0.05, P adjusted value 0.1; edgerR P-value 0.05,
FDR 0.1. The parameters are those used in point 4.2 so that the
results can be compared.
The final result is divided into some innovative cornerstones:
from the 28 overexpressed genes (23 in the luminal and 5 in the
nonluminal), we found that
1. DSCAM-AS1 is a specific gene of the luminal A subtype that is
not present in healthy tissue or preneoplastic lesions. If this
gene is present, the patient could avoid standard PBI and
adjuvant therapy.
2. ER+ patients (good prognosis) died because of a high risk for
the overexpression of CCND1, INST4, and GAB2.
3. ER+ patients (good prognosis) died because the overexpression
of FOXJ3, SOX2, and FGFR3 (4 times less expressed in the
luminal region) correlated with the failure of endocrine
therapy.
4. in HERB2+ patients, we found other genes, among which
PAX8-AS1 was responsible for stem cell proliferation.
5. The fusion of IFITM10 and CTSD promotes cell proliferation of
the tumor and is a tumor marker.
The study highlighted a code of 33 genes that characterize
breast cancer. Forty-three percent of the isolated genes were
common between the 1st and 2nd sequences. Seven of our genes
are found in the commercial genetic tests PAM50, Oncotype and
Endopredict: four are present in the Panel Cancer targets 50 genes
of the Ion AmpliSeq. The verification of the validation of the 28 genes
selected from phase I in a more recent sample of women (2005) and
characterized by the molecular split point (Endopredict®) allowed
us to provide the study with greater consistency (Table 2).
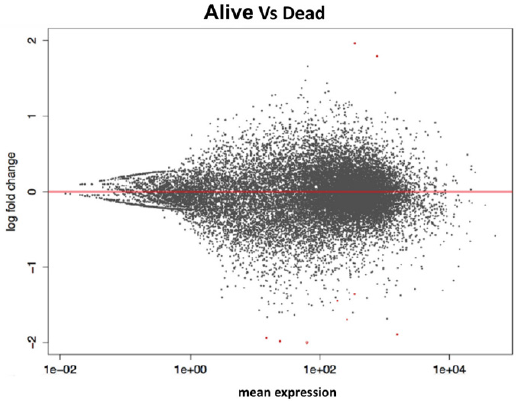
Figure 2: Life vs Dead.
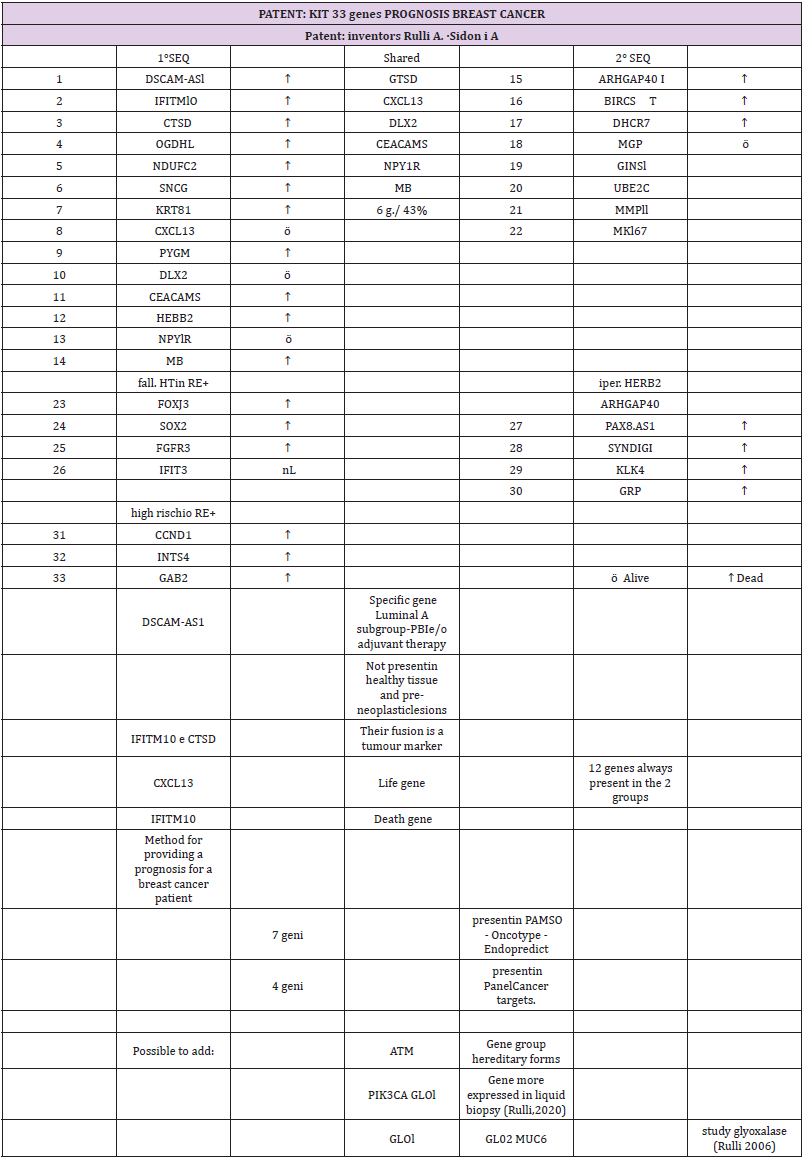
Table 2: The 28 genes characterized by the molecular split point (Endopredict®).
Conclusion
A very positive result was the ability to extract suitable RNA
from 1994 samples in paraffin in good quantities and qualities to
make the study possible. In this regard, experiments have been
conducted to define the best protocol between RNA extraction
and library preparation. The calculation of the RNA (RNA integrity
number) and the concentration at Qubit allowed us to always
“select” valid samples. The basic hypothesis of the study has been
confirmed: the characterization of the luminal and nonluminal
tumors is not real through IHC (surrogate St. Gallen), which is
routinely used but must be read on a molecular basis. However,
the most relevant data are represented by the 33 overexpressed
genes: 28 genes (23 in the luminal and 5 in the nonluminal) and 5
genes (which confirm the premises of the study in wanting to find
molecular markers capable of “personalizing” the therapy). Two of
the 28 genes were always present in both groups: a CXCL13 life gene
and an IFITM10 death gene. Moreover, IFITM10 and CTSD fusion
promotes cell proliferation of the tumor and is a tumor marker. The
DSCAM-AS1 gene is specific to the luminal A subtype. If this gene is
present, the patient could avoid standard PBI and adjuvant therapy.
The result obtained, which can be assumed to be transformed
into a genetic panel, following validation on more recent samples
(2005) and studied with Endopredict®, will help us to implement
a personalization of the therapy: surgical and adjuvant. In the
preoperative phase, with the core biopsy of the neoplasm, the
histological diagnosis is obtained, and then the biopathological
characterization and the presence of the genes described above are
verified to evaluate the risk of local and/or systemic recurrence,
which varies according to the molecular subtype.The hyperexpression of CCND1, INST4 and GAB2 changes the
prognosis of ER+ patients from favorable to inaustic. Our work also
revealed that ER+ patients died (in which we would have expected a
good prognosis) because they had overexpressed FOXJ3, SOX2, and
FGFR3 genes that correlate with the failure of endocrine therapy.
In the postoperative phase, targeted therapy allows a better
stratification of adjuvant therapies based on the amplification of
the genes that regulate, for example, resistance to tamoxifen and/
or to trastuzumab.
For more Articles on : https://biomedres01.blogspot.com/



No comments:
Post a Comment
Note: Only a member of this blog may post a comment.