The Current State of Target Therapy for Subtypes of Gastrointestinal Stromal Tumors
Introduction
Gastrointestinal stromal tumors (GISTs) are the most common
primary mesenchymal tumor of the gastrointestinal tract, although
with low incidence. They are connective tissue tumor arising from
the interstitial cells of Cajal (ICC) cells. The most common tumor
location is the stomach (50%-70%), followed by the small intestine
(25%-35%), rectum (5%-10%), and esophagus (<5%) [1]. The
signs and symptoms of GISTs, which grow slowly and generally
occur in 50 to 70 years old, are gastrointestinal hemorrhage,
trouble in swallowing and metastases [2]. About 30% of GISTs can
be defined as evil and led to metastasize occurring drug resistance
during the treatment because of gene mutuation [3]. Some study
show that the patients of GIST have mutations in PDGFR (PDGFRα
or PDGFRA) but not in the wild-type GISTs without c-kit or PDGFRA
mutations were accounted for 10-15% of the total number of
GISTs (Table 1) [4] According to researchs, wild-type GISTs might
lack succinate dehydrogenase (SDH) leading by mutations of SDH genes.The occurrence of GISTs are related with KIT/PDGFRA gene
mutations, which c-kit mutations were accounted for 80~90% and
PDGFRA mutations for 7% [5].
The radical surgery may be the only chance of cure, but the treatments of traditional radiation and chemotherapy are ineffective for advanced GIST (recurrent, inoperable or distant metastasis). In recent years, the targeted therapy of GISTs has obtained significant efficacy with the development of precision medical. Tyrosine kinase inhibitor (TKI) is the first-line treatment drug of GISTs, such as imatinib mesylate (IM). The locus secondary mutations of KIT/PDGFRA genes were key factors for prognosis. At the same time, the application of targeted drugs to achieve the maximum therapeutic effect has become the focus of research. The latest NIH classification system for GISTs declare that Mid- to Late GISTs are applicative for the adjuvant therapy, which preoperative IM is widely available. This article aims to make a summary of different genotype, mutation and molecular mechanism of GISTs, and the progress of targeted drugs.

Table 1: Genetic mutation in GIST.
Genetic Mutation in GISTs
KIT Mutation
C-kit gene is considered at the homologue of HZ4 feline sarcoma
Virus KITs oncogene. C-kit proto-oncogene, which consisting of
21 coding exons in III type of protein tyrosine kinase receptor
superfamily members, is located in chromosome 4q11-12. It is a
kind of Kit protein receptor, which consist of extra-cellular region,
trans-membrane region, near-membrane region and 2 tyrosine
kinase (TK) region. Its ligand is stem cell growth factor (SCF). The
most common mutations are exon11 mutations (70%), exon9
mutations (5%~10%), and exon13 mutations (1%) [6]. Exon14,
17, 18 mutations are relatively rare [7]. The main mutation types
are deletion mutation, point mutation, mixed mutation and insert
mutation [8].
When c-kit mutated, CD117 protein expresses and forms a dimer
autonomously without the participation of the ligand. Therefore,
it cannot precisely regulate the differentiation, proliferation, and
programmed cell death. Finally, c-kit mutations can cause more
cells enter the neoplastic hyperplasia stage from the quiescent
stage, which may be one of the key mechanisms causing malignant
transformation of GIST. Studies have found that tumors with c-kit
exon11 deletion are more likely to appear in the patients who are
older than 50 years, with a tumor diameter of 5~10cm and the NIH
grade as high risk. Moreover, the secondary and high-risk GISTs
are susceptible to secondary mutations that cause recurrence and
metastasis because of the deletion of exon11 of c-kit gene [9], which
has potential value for predicting poor prognosis of patients.
PDGFRA Mutation
PDGFRA gene stretches approximately 65 kb on human chromosome 4 (4, q11-13) and consists of 23 exons in III type of protein tyrosine kinase receptor superfamily members, including exon3-10 coding region exocellular five immunoglobulin sample, exon11 coding intracellular membrane area, near exon13-15 and exon17-21 coding intracellular kinase 2 cheese ammonia acid (tyrosine kinase, TK) area. Similar to c-kit, PDGFRA gene mutation is also a function-acquired mutation and most of them occur in CD117 negative GISTs [10]. In the absence of ligand binding, the autonomous dimerization leads to cell division and proliferation by promoting DNA synthesis through downstream signaling pathways, thus the growth, proliferation, attachment, metastasis, differentiation, and apoptosis of cells are regulated. The mutations of PDGFRA essentially clustered in three regions and the most common of these is exon18 that accounting for 82.5%, the most common site is D842V [11-13]. The process of GISTs formation by PDGFRA mutation is similar to c-Kit, the tyrosine kinase receptor encoded by PDGFRA located on the cell membrane is abnormally and continuously activated, resulting in the loss of autoinhibition function.
Wild-Type GIST
The morphology of wild type GIST is in line with the GIST,
CD117 positive or negative expression, at the same time cannot
detect c-kit and PDGFRA gene mutations, but often coupled with
abnormalities in the structure or expression of other genes, such
as, subunits of succinate dehydrogenase (SDH) complex, Rapidly
accelerated fubrosarcoma B (BRAF) gene, neurofibromatosistype1
(NF1) gene mutation, multiple gene fusion and other abnormalities.
The most common type in the wild type is the SDH expression
deficient or decreased, which can be accompanied by other
neoplastic diseases, including paraganglioma, pulmonary
chondroma, pheochromocytoma, etc. At first, the disease was
recognized as Carney triad and Carney Stratakis syndrome [14].
The high expression of insulin-like growth factor 1 receptor
(IGF-1R) receptor RNA or protein in the GISTs due to SDH gene
defects is detected,IGF-1R binds to ligand and is activated by
autophosphorylation, leading to the activation of mitogen-activated
protein kinase (MAPK) and phosphatidlinositol 3-kinase (PI3K)
cascades [15].
IGF signaling can inhibit IGF-1R-induced apoptosis in SDHdeficient
GIST cells and inhibit the signaling of Akt and MAPK
pathways in GIST cells [16]. For BRAF gene mutant GIST, most
of the mutation sites are V600E of exon 15, which can affect the
function of PI3K [17], BRAF mutations have been found in a small
number of patients with imatinib (IM) resistance, suggesting that
BRAF mutations may be the cause of secondary resistance [18].
KRAS mutations are also one of the wild-type GIST, some scholars
analyzed the gene sequence of 578 cases of GIST and found no KRAS
mutation, So KRAS mutations may be extremely rare [19].
Other Biological Markers
Many research experiments show that c-kit mutations represent a poor prognosis in high-risk GISTS, PDGFRA mutations associated with less malignant GIST [20]. However, these alone are not absolute and require more biological indicators related to prognosis and efficacy. In addition, the proliferation index Ki-67 is a very effective marker for predicting the aggressive behavior and malignant potential of GIST and can be employed as an independent predictor of GIST [21]. P16 expression is also related to the malignant of GIST [22]. Moreover, there are evidence from 19 studies showing p53 have the predictive value in the risk of GIST [23].
TKI Therapy in GIST
Complete surgical resection is the only chance for cure for the disease, but there is still a possibility of recurrence and metastasis. For advanced (unresectable or recurrent, metastatic) GISTs, molecular targeted therapy is the preferred treatment [24].
First-Line Imatinib Therapy for Advanced GIST
Imatinib is a derivative of 2-phenylaminopyrimidine, which
is a kind of small molecule selectively activated tyrosine enzyme
inhibition agent to inhibit the BCR-ABL protein, ABL, KIT and
PDGFRs. Though selective inhibition of tyrosine kinase lives sex,
blocking the phosphate group on tyrosine residues. In 2002, the
FDA approved STI571 for the treatment of unresected or metastatic
GIST. Demitri et al conducted a trial, 147 patients with advanced
GIST were randomly assigned to receive 400mg and 600mg of
imatinib per day. The results showed that imatinib has achieved
a good objective response in patients with advanced GIST, and
there were no significant differences in the safety of treatment
dose [25]. Subsequent phase 2 and phase 3 trials of metastatic
GIST also confirmed the efficacy of imatinib in advanced GIST
(Table 2). Treatments which provided the sites to inhibit tumor cell
proliferation were widely applied advanced and metastatic tumor,
and the preoperative and postoperative adjuvant therapy.
However, an effective dose of imatinib cannot inhibit other open
sites in the tyrosine kinase pathway except for c-kit or PDGFRA
gene mutations, allowing cell proliferation signals to bypass cell
proliferation inhibition caused by imatinib, c -kit exon11 mutant
GIST is generally sensitive to imatinib and not sensitive to exon9
and PDGFRA mutation types [26]. Further research found that the
relationship between kinase genotype and treatment outcome,
Kit exon11 genotype mutation has better benefit than other
genotype mutations [27]. Our previous meta-analysis estimated
the imatinib treatment for different genotypes of GIST and found
that personalized treatment makes patients with exon 11 mutation
more profitable [28]. To optimize the treatment of imatinib, a metaanalysis
of 1,640 patients with advanced disease showed that the
patient could get a small PFS advantage of 2 times the standard
dose (800mg/d) of imatinib, but no significant difference in OS
between the dosages, especially in kit exon9 mutation.
Compared with exon11 mutation, exon9 mutation and wild
mutation have a worse prognosis [29]. Moreover, Asian Consensus
Guidelines agreed that a higher dose may also be beneficial in
Asian patients with KIT exon 9 mutation [30]. To assess longterm
survival with two doses of imatinib, one long-term result of
a randomized trial showed the 800mg/d group had a better 10-
year PFS rate and an average OS rate than the 400mg/d group [31].
Whether patients with advanced GIST benefit significantly in terms
of long-term survival and will they differ due to the differences of
mutations? An analysis of Phase 3 SWOG Intergroup Trial S0033
showed that imatinib as a first-line drug in advanced GIST has
resulted in long-term survival of 10 years for a significant number
of patients, especially those with KIT exon11 mutations or KIT/
PDGFRA mutations lacking (mainly succinic dehydrogenase mutant
tumors) [32]. Most people can benefit from imatinib, but this result
does not lasting. Many patients will initially be resistant even if they
were recommended to extend the duration of medication from one
year to three years [20].
GIST resistance of imatinib can be divided into primary
and secondary, primary resistance is no response to imatinib
treatment, while secondary resistance occurs 6 months after the
initial treatment is effective. The secondary gene mutation of KIT/
PDGFRA is considered closely related to secondary resistance.
The secondary mutation of kit occurred in exons 13, 17, 14 [33]
and occurred in exon 18 of PDGFRA [34]. Further research found
that heterogeneity of KIT secondary mutations is the principal
mechanism of tumour progression to KIT inhibitors in imatinibresistant
GIST patients [35]. The sensitivity of PDGFRA mutant
GISTs to targeted drugs is obviously worse than that of C-KIT,
while the exon 18 D842V mutation of PDGFRA mutant GISTs
is also primary resistance to imatinib, so it is difficult to achieve
satisfactory efficacy even if the dose of imatinib is increased [36].
Many resistance mechanisms are still being researched due to the
diversity of mutation sites, heterogeneous multitarget inhibitors
and precise personal treatment are more worthy of promotion.
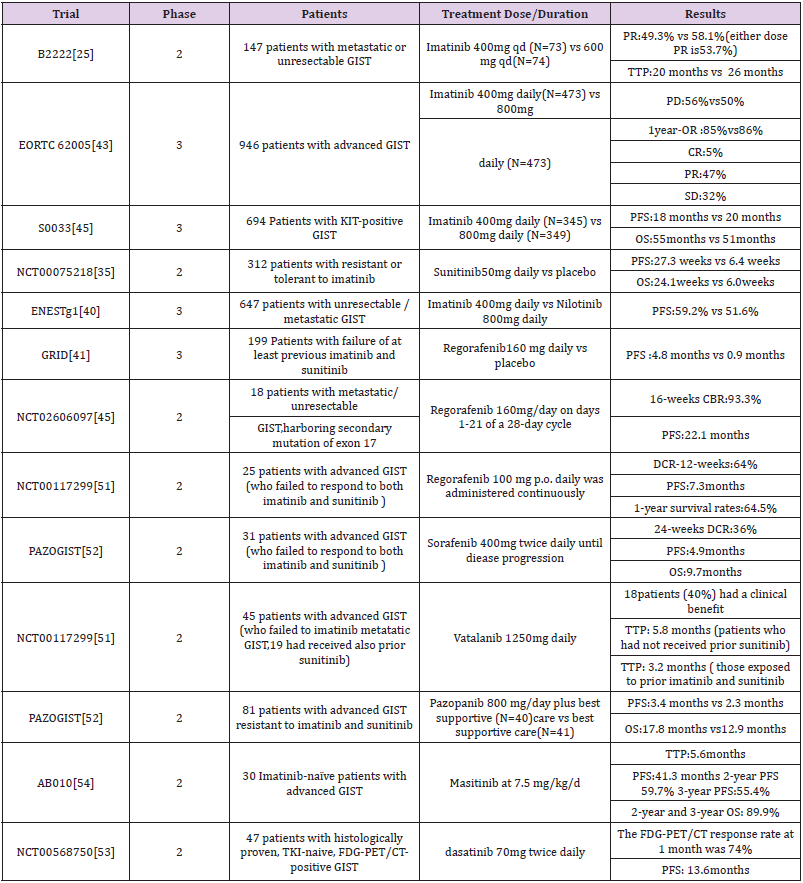
Table 2: Some important trials about TKIs.
Second-line Sunitinib for Advanced GIST
Secondary drug resistance occurred in more than 50% of patients after treatment with imatinib, thus the second-line treatment drugs such as sunitinib, an oral multitarget tyrosinase inhibitor, emerged. The growth, proliferation and metastasis of malignant tumors were affected by blocking the signaling pathway by inhibiting tyrosine kinases such as VEGFR, PDGFR, KIT, and RET. Sunitinib is considered as a second line TKI because of its considerable benefit in patients with advanced imatinib resistance and intolerance. A randomized, blank-control phase 2 clinical trial of random taking sunitinib 50mg / d and placebo in the blank control group. The results of this experiment showed that progressionfree survivals were 27.3 weeks and 6.4 weeks, respectively. Overall survival was 24.1 weeks and 6 weeks, respectively (Table 2) [37]. However, Lile Wu et al. conducted a meta-analysis clinical efficacy of seconed-generation TKI in imatinib-resisitant GISTS showed that sunitinib are effective for improving PFS but not OS in patients with imatinib-resistant GIST [38].
One of our previous pooled analysis showed that sunitinib treatment after imatinib resistance varied according to the genetic subtypes of GIST. Compared with PDGFRA, the mutation of KIT gene showed better clinical rate of cure, especially the mutation of KIT exon 9, 11. Furthermore, the mutation cure rate of KIT exon 9 was better than that of exon 11 [39]. Other second-line TKI drugs such as nilotinib, a tyrosine kinase activity of ABL/BCR, and KIT, PDGFRs, significantly improve PFS [40]. In a phase III study of nilotinib versus imatinib as first-line therapy for unresectable or metastatic GIST, Blay JY et al. divided 324 advanced patients on nilotinib 400 mg twice daily and 320 advanced patients on imatinib 400 mg daily. The results showed that the 2-year progression-free survival rate was better in the imatinib group (59.2%) than in the nilotinib group (51.6%). There was no difference between the two groups for progression-free survival of KIT exon 11 mutation, but imatinib group was better than nilotinib of the exon 9 mutation. Therefore, in the future, first-line drugs need to be determined based on subtype analysis [41].
Third Line Regorafeniband Ripretinib for Advanced GIST
Regorafenib, a multikinase inhibitor was candidated as a
third-line treatment option for patients with advanced GIST,
which is recommended after failure of both high-dose imatinib
and sunitinib. In a randomized phaseIII trial of regorafenib GRID,
the results showed that median progression-free survival was
4.8 months for regorafenib and 0.9 months for placebo, with no
significant difference in overall survival [42]. Another study of
regorafenib showed the efficacy and safety of manageable profile
in Japanese were consistent with the overall GRID study population
of patients with advanced GIST. As a third line TKI, the effectiveness
of regorafenib has been proven in GIRD, however, frequent dose
reductions were required of the administration plan. In a study
involving 25 patients who failed treatment with imatinib and
sunitinib, low-dose continuous treatment with regorafenib showed
that the disease control rate for at least 3 months in 64% of patients
and had a median progression-free survival of 7.3 months. Maybe
this method of administration has become a better choice [43,44].
In addition, regorafenib prolonged progression-free survival of
patients with advanced mutations in exon17 [45-48]. Another threeline
targeted therapy, ripretinib is a Ⅱ type switchpocket inhibitor
that binds to the switchpocket and acts as a structural substitute for
an inhibitory switch, preventing the activation loop from entering
the switchpocket, thus locking the kinase in an inactive state and
inhibiting downstream signaling. Ripretinib has strong inhibitory
effects on different secondary drug resistance mutations because it
is acting on the final link of the kinase pathway [49-50]. However,
clinical data on ripretinib therapy and genotyping have not been
reported yet, so there may be differences in the efficacy of ripretinib
in GIST with different genetic mutation types.
Other TKIs
Other TKIs identified in clinical trials, such as sorafenib, as a third line / four-line TKI. A Korean clinical trial used two or more TKIs to treat unresectable or distant metastic, 36% of patients had more than six months of disease control after using sorafenib [51]. Moreover, including vatalanib (PTK789), masatinib (AB1010), pazopanib (PAZOGIST), dasatinib (NCT0056875) (Table 2) [52-54].
Conclusion
Despite the many benefits of targeted drugs for advanced
GIST, drug resistance presents a new challenge. Through the
understanding of the mutation sites of GIST and the collection of
information about current treatment regimens, we can see that
the efficacy of targeted drugs in different genotypes still needs
a large number of standardized clinical open trials. Similarly,
to leverage the strength of targeted drugs to make up for the
shortcomings, personalized treatment and evaluation are essential.
New therapeutic ideas should be applied to more actions, such as
the combination of targeted and chemotherapy drugs, new targets
to be discovered, updated TKIs and combined immunotherapy.
Further studies on drug resistance mechanisms and new molecular
markers are the breakthrough points for the treatment of advanced
GIST to optimize the treatment of advanced GIST.
For more Articles on : https://biomedres01.blogspot.com/



No comments:
Post a Comment
Note: Only a member of this blog may post a comment.