Magnetc Torque in Superoxide Ion is the Main Driving Force of Dioxygen Activation in Aerobic Life
Introduction
Oxygen produced by photosynthesis is everywhere - in air, in
water and inside all aerobic creatures including ourselves. It plays
such a great role in all biosphere that it is called “a wonderful elixir
of life from the green leafs under the Sun” [1]. The main factor,
which determines all wonderful properties of oxygen being the
origin of aerobic life on the Earth can be expressed in one word and
this word is spin. Electronic spin of the oxygen molecule explains
the origin of all paradoxes in physics and chemistry of this simple
diatomic species including its importance for living matter [1-
19]. At the same time the well-known chemical concepts which
generalize the role of electron spin in chemical reactions [14] are
not fully applied for bio-oxidation processes [1]. Respiration and
combustion are known to be equivalent in the balance of their
exothermicity and the yield of reaction products; all carbohydrates
and hydro-carbons are oxidized by molecular oxygen to H2O and
CO2. Ignition of organic fuel in the open air provides occurrence of the
first radicals, which can initiate a radical-chain process thereby
removing the spin-forbidden character of O2 reaction with organic
substances. Such type of dioxygen activation is impossible in-side
the living cell since the radicals would burn the cell.
Aerobic organisms use special enzymes, which involve
magnetic perturbations affording to induce ISC and brake the
severe spin selection rule for two unpaired electrons of dioxygen
during bio-oxidation of organic matter; that is to overcome spin
selection and sever quantum prohibition for triplet O2 biradical to
react with diamagnetic organic substances. Magnetic spin catalysis
of concerted reactions of molecular oxygen in the enzyme active
center does not correspond to the classical chemical concepts and
common organic reaction rules. One has to stress that only magnetic
forces are able to induce spin flip, which separates the unpaired O2
electrons from the diamagnetic products of bio-oxidation (CO2, N2
and water molecules with all spins paired). Internal magnetic forces
in molecules are much weaker than electric forces which govern
all main chemical interactions; bond strength, heat of formation,
dissociation and activation energy accompanied by the spin flip can
be considered only with relativistic quantum chemistry methods.
These quantum processes inside living matter do not obey the
common chemistry rules and are determined by internal magnetic
interactions being specific to the electronic open shell of dioxygen.
They are important for all aerobic evolution, photo-synthesis,
appearance of reactive oxygen species and superoxide dismutase,
oxi-dative stress and signalling functions in plants.
The problem of molecular oxygen activation is a longstanding
task in bio-chemistry [1-6]. In contrast to the majority
of stable organic substances, M in Eq. (1), which usually are dia
magnetics, since their electron spins are paired and the total spin
quantum number (S) is zero, the molecular oxygen (dioxygen) is
a paramagnetic gas. That is, the O2 molecule has a ground triplet
state X3Σg with a nonzero electron spin (S=1) produced by two nonpaired
electrons at the degenerate highest occupied molecular
orbital (HOMO) πg [1]. The presence of spin in the ground state
of O2 leads to a strict quantum prohibition on oxidation of organic
substrates by dioxygen, since the reaction products (P = CO2, H2O,
N2) are also diamagnetic species.
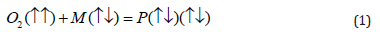
The M molecule has a singlet ground state (S=0) and the total spin of reactants is triplet (S=1), while the product has a singlet state again. Thus, combustion of organic matter is strictly spinforbidden if no special initiation by additional radical R, which is responsible for ignition.
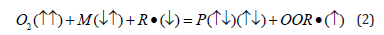
The total spin (S=1/2) and its projection on z-axis (Ms=+1/2)
are the same in the left and right part of Eq. (2). Thus, the radicalinitiated
combustion proceeds as radical-chain reaction until the
whole fuel M would be exhausted or the chain-transfer radical
would be saturated and removed by a scavenger [1]. In the absence
of radicals the direct oxidation by dioxygen is spin-forbidden, Eq.
(1); this is the reason why our world had not been burnt when
photosynthesis starts to saturate the early Earth’s atmosphere and
primordial oceans. The newborn radicals OOR• can continue the
chain reaction with dioxygen without spin prohibition. Reaction
from Eq. (2) can be repeated with any peroxide or other available
radical R• [14]. Usually, this is a branched chain reaction which
provides exothermic effect in the form of flame. In the flame there
are many various radicals and random collisions are possible in
the plasma volume; the newborn radical, Eq. (2), escapes into the
volume and its spin orientation is arbitrary for new collisions. Thus,
it loses spin memory, which was created during radical appearance,
Eq. (2). In a new subsequent collision with new O2 molecule a
random spin orientation can be realized, since the former spin
memory is lost. But it is not the case for biooxidation inside the
cellular enzyme where all possible radicals, if any are born, cannot
leave the active center; they keep their spin memory obtained
during the birth moment. The “fiery” mechanism, Eq. (2), in the
cell is impossible also because the energy supply cannot be proper
regulated in radical-chain reaction.
At the same time, the spin selection determined by the triplet
ground state of dioxygen leads also to forbidden optical transitions
in the visible and near IR spectrum of the O2 molecule [9-16]. By
that reason our atmosphere is transparent to visible sun light,
though the energies of the singlet excited states of dioxygen
just correspond to the red and near infrared (NIR) range of
electromagnetic waves. The singlet  oxygen (known as one
of the most reactive oxygen species in the oxidative stress) is
monitored now in tissues by its NIR luminescence at 1.27μm [9-12].
Analysis of dioxygen wave-functions and transition moments in the
electronic O2 spectrum sheds light on the role of internal magnetic
forces, which make it possible to overcome spin restrictions in both
enigmatic phenomena - in light emission and in biological oxidation
by dioxygen [1,12].
oxygen (known as one
of the most reactive oxygen species in the oxidative stress) is
monitored now in tissues by its NIR luminescence at 1.27μm [9-12].
Analysis of dioxygen wave-functions and transition moments in the
electronic O2 spectrum sheds light on the role of internal magnetic
forces, which make it possible to overcome spin restrictions in both
enigmatic phenomena - in light emission and in biological oxidation
by dioxygen [1,12].
Oxidizing Power of Dioxygen and its Slaggish Reactivity without Enzymes
We used to take our respiration for granted and do not care much about spin of dioxygen; although, we have to care. The high reduction potential of dioxygen causes its great oxidizing power [3]. Using only thermodynamic backgrounds one can predict that O2 is one of the best oxidizing agent [7]. But the triplet spin in the ground state of dioxygen prevents spontaneous oxidation of organic matter and its full combustion into puf of smoke and soot. Respiration of aerobic creatures consumes almost 90% of all dioxygen produced by photosynthesis and utilized in the Earth’s biosphere [6,19]. In aerobic organisms about 80% of the consumed dioxygen is utilized during conversion of food (glucose) to the viable ATP energy sources. This oxidative phosphorilation is catalyzed finally by cytochrome c oxidase in most aerobic organisms [5,18]. This terminal step of respiration represents the four-electron reduction of dioxygen in order to produce two water molecules, Eq. 3 [5,6]:

The overall resulting reduction potential of reaction (3) is equal
+0.815 V versus NHE (normal hydrogen electrode); this means that
such reduction is highly favored in terms of thermodynamics [5,13-
20]. Much of the rest of the consumed dioxygen besides respiration,
Eq. 3, is utilized during metabolism for biosynthesis of proteins and
other useful molecular substrates. This biosynthesis is catalyzed
by monooxygenases or dioxygenases. With these enzymes one or
two oxygen atoms from dioxygen molecule are incorporated into
the final products, respectively [17,18]. The overall reduction, Eq.
(3), usually proceeds through a series of one and two electron
transfer steps [2-5]. These reactions are strongly favored by
reduction potentials expecting the one-electron reduction of O2
to produce anion-radical superoxide 2 O •− [5,19]. The late process
has a reduction potential equals -0.33V versus NHE; this relatively
low reduction potential for one-electron transfer to dioxygen is
an important factors which limits the kinetic reactivity of O2 [5].
But this is not the most important factor! The point is that the high
oxidizing power stored up in O2 molecule cannot be realized until
the first one-electron jump and reduction to O2
•- will be produced.
And the following reaction step in the enzyme active center has to
proceed in a concerted manner with the same reducing agent E,
which is responsible for electron transfer to O2. This means that the
radical pair (RP) E+•--- O2
•- cannot leave the enzyme active center
and the RP cannot dissociate before the spin-forbidden reaction
in the triplet RP will occur [1-3]. The starting RP (E+•--- O2
•-) has
the triplet ground state since the precursors (E + O2) have two
non-paired spins in dioxygen moiety and the total spin S=1. Spin
flip and triplet singlet (T-S) transition cannot be realized during
electron transfer since the spin-conserving reduction step is much
faster anywhere [13]. The necessary T-S transition can occur only at
the geminate triplet radical pair step and this review is devoted to
prove this statement.
The main paradox of life is that aerobic organisms cannot
receive enough energy and survive without dioxygen, but at the
same time O2 from the air is quite dangerous to their existence
because of generation of reactive oxygen species (ROS) as
byproducts of oxidative metabolism. This contradictory and
inherent property of dioxygen relates to the triplet ground state
of O2 molecule with the total spin (S) quantum number S=1. This
quantum number determines expectation value of the S2 operator,
where S = S1 + S2 is the total spin angular momentum for two
electrons. Each electron has spin quantum number S= ½ and two
projection on the chosen z-axis ms = ± 1/2. The absolute value
of |S| or a length of the spin angular momentum vector of one
electron is equal to 0.866ℏ. This value determines a strength of its
magnetic field, which was measured and confirmed in numerous
experiments [14]. For the triplet state Ms = ± 1, 0 and we have three
possible projection of the spin angular momentum on molecular
axis in the O2 molecule. Not many biochemists had payed proper
attention to this fundamental property of dioxygen, especially to
its sequences for respiration and oxidative stress. Both mysterious
phenomena are determined by magnetic perturbations in the πg
open shell of dioxygen [1,14]. Thus, O2 is a stable free biradical
and its reactions with organic molecules (which have the singlet
ground state, S=0) are forbidden by severe spin selection rule.
By this reason, dioxygen shows very sluggish chemical reactivity
at ambient conditions (in the absence of radicals with unpaired
electron spin) in spite of its strong thermodynamic potential for
oxidation. This obstacle explains the second paradox of dioxygen
reactivity; the high exothermicity of organic material oxygenation
is in a great contradiction with a low reaction rate of their reactions
with dioxygen in the absence of radicals [1]. According to the Polani
Semenov rule the high exothermic effect of reaction correlates with
its low activation barrier and the high reaction rate constant; the O2
reactivity demonstrates an opposite behavior [1]. Spin prohibition
does not permit to realize the high oxidation power of dioxygen in
terms of normal chemical kinetics [3]. That is why our world had
not consumed into fire when green-blue algae started to saturate
the Earth’s atmosphere by photosynthetic dioxygen 2.4 billions
years ago (BYA). That time is known as the Great Oxygen Event
(GOE) or Oxygen Catastrophe, when anaerobic forms of life and
archaea perished and were substituted by eukariots; thus, a new
efficient type of aerobic evolution had started since then [8]. Before
GOE the primordial atmosphere does not contain, or contains very
little dioxygen. The notion of GOE is deeply imprinted now in our
understanding of biosphere on the early Earth and of great role of
this ecological catastrophe which ever shaked our planet. The impact
of dioxygen spin state that time was much more important for
living archaea than this spin is conceivable nowadays for the whole
human civilization of man-kind.
A chronology of dioxygen accumulation on the Earth is rather
complicated and includes about 900 million years from 3.3 to
2.4 BYA [20]. The increase of O2 concentration in primordial
atmosphere, from 0.1% up to 15% during GOE, provided finally
a great change in biochemical reactions responsible for energy
supply from simple glycolysis in anaerobic bacteria to oxidative
phosphorilation (OP) in eukariots as well as a new opportunity
for cell proliferation and biological diversification. Anaerobic life
based on glicolysis had been rather limited energetically (despite
its efficient recycling of organic matter); only two ATP molecules
are produced during glucose conversion into two molecules of pyruvate in
the absence of O2 [5,6]. Glycolysis still exists and takes
place in the cytosol of modern eukariots in some emergency cases.
But it was the only source of energy until the GOE time when O2
gas became widespread available on the Earth in air and oceans.
The tricarboxylic acid (Krebs) cycle using dioxyen provides 32
ATP molecules, instead of two. Thus, much more free energy of
adenosine triphosphate (ATP) became available to plants and
animals and provided a breakthrough in evolution [5]. The advent
of triplet dioxygen increased aerobic metabolism dramatically and
made global impacts on all environment. What was the role of O2
electron spin in this Great Oxidation Event? Let us try to collect first
the well-known facts.
One can consider mitochondria created and evolved after the
GOE [5]. These mitochondria gave to aerobic cells much more
energy exploiting oxidative phosphorilation with new and more
complex morphology of inner membranes [5]. These are the places
of the tricarboxylic acid cycle and the OP realization by which a
large number of ATP molecules are produced from organic fuel.
This is done through the electrochemical proton gradient, which
is generated across the inner membranes by the high-energy
electrons, which are passing along the electron transport chain
[6]. But the role of mitochondria in living organisms extends far
beyond oxidation of glucose via oxidative phosphorilation; it
includes synthesis of haem, hormones, amino acids and many other
molecules used for metabolism [7,18]. The recently discovered
new role of mitochondria is their involvement in apoptosis and
ion homeostasis through the signaling functions of ROS [8]. During
the OP process mitochondria utilize dioxygen to generate ATP,
but conjugation of this process with the electron transport chain
can lead to O2 partial reduction and to superoxide anion (O2
•--)
generation. This is the most important reactive oxygen species
being the ancestor of other ROS, which are partly reduced forms
of dioxygen: the hydrogen peroxide (H2O2), very reactive hydroxyl
(OH•), peroxyl radicals (ROO•) and the singlet 1
2 g O Δ dioxygen [8].
Highly likely, that ROS appeared on the Earth during GOE together
with the first photosynthetic dioxygen and ROS have been a
permanent companion of aerobic evolution ever since [8].
In addition, one can take into account the highly-reducing
milleu and high lev-el of reduced iron in the ancient oceans which
provide efficient conversion of the first photosynthetic dioxygen
into ROS [8]; probably, by this reason the former anaerobic life had
perished. Dioxygen reduction in that milleu produced superoxide
anion, which triggered other processes of ROS proliferation. The
O2
•-- anion dismutates to form H2O2, which interacts with the soluble
Fe(II) ions by the Fen-ton reaction [21] to form reactive hydroxyl
radical and life became very dangerous. Superoxide dismutase
(SOD), the oldest enzyme that scavenges ROS effectively was
found in all kingdoms of life; moreover, SOD has been evolved even
before the eubacteria differentiation from archaea [8]. The GOE
left many finger prints in the rocks and those records clearly show
the early Earth’s evolution [20]. The finding of old history of SOD
strongly approves the simultaneous ROS appearance together with
photosynthesis, dioxygen and first oxidative enzymes. Considering
the current role of ROS in modern plants and mammals together
with dioxygen activation mechanisms, one has to keep in mind
that aerobic evolution on the Earth occurred in the presence of
ROS and increasing O2 concentration. The first primitive organisms
have finally evolved the strategy to survive and cope with the
abundant dioxygen in the GOE-growing atmosphere. The O2-
utilizing enzymes started to use paramagnetic metal ions, which
provided spin-crossover in their d-shell assisting the T-S transition
in dioxygen moiety during re-dox reactions with substrates [1-
3,14]. This type of spin catalysis is rather simple and determined
by exchange interactions [3]. Formally, catalysis by metal cofactor
is allowed by spin-selection rules and its efficiency depends on the
differences in exchange integrals for different electron pairs in the
transition state region Otherwise, the evolved metal-free cofactors
are more complicated in terms of their spin dynamic mechanisms.
These spin transformations are met in numerous flavoenzymes
[2,19]. Flavins and related pterins are extremely miscellaneous and
versatile cofactors, which capable to activate O2 [18].
Oxidases and Monooxygenases with Flavin Cofactors
Now we want to consider the role of electron spin and internal
magnetic interactions in the dioxygen open-shell, which are
responsible for O2 activation by flavin-containing enzymes. This
very wide class of catalysts provides numerous oxidation and
oxygenation reactions in living cells. Some general results from
such consideration will lead to useful conclusions about aerobic
evolution and the role of dioxygen spin in our life.
Flavoenzymes as a widespread class of proteins are more
adjusted to O2 activation than free flavins (Figure 1). Besides their
enabling for efficient oxidations of the target substrates by triplet
dioxygen they can supply the reduced reactive oxygen species
to arrange signaling network system in the cell. ROS have been
referred long time as “the double-edged sword of life” because of
their toxic nature but al-so of their beneficial role [8]. The modern
trends stress the great importance of the late role especially for
plants [8,18]. Physical, chemical and biological principles by
which flavoenzymes realize catalytic action upon dioxygen have
been the subject of intense studies during last sixty years [5-7,18].
Current understanding of these rules is based mostly on seminal
works of Vincent Massey [7,19]. A wealth of kinetic, mechanistic,
computational and spectroscopy data [7] represent his consistent
view on the functional and structural properties of flavin-containing
oxidases and oxygenases. However, not all aspects of flavoproteincatalyzed
bioreactions have been solved yet [18]. The major area of the flavoenzyme research still concentrates on elucidation of the
electronic mechanisms and molecular basis for the dioxygen spin
activation [1,22].
Nowadays, more than 105 protein sequences deposited in the
protein database are considered as flavin-dependent enzymes
[18]. Flavoenzyme cofactors usually include either flavin adenine
dinucleotide (FAD) or flavin mononucleotide (FMN), both being
synthesized from vitamin B2 in vivo (Figure 1). The O2 spinactivating
mechanism of flavoenzymes was first proposed for
oxidases [2]. For example, glucose oxidase (GO) utilizes dioxygen as
an electron acceptor in order to produce hydrogen peroxide [2-5].
Similar spin flip occurs when one oxygen atoms from O2 molecule
is incorporating into the products; in this way monooxygenases
activate dioxygen through spin catalysis by forming the C(4a)
peroxyflavin intermediate, which then reacts with substrate and
inserts one oxygen atom into substrate by ordinary chemical
rearrangement [18,22]. Free reduced flavins also react with
dioxygen in solvents quite efficiently, forming oxidized flavin and
hydrogen peroxide faster than in one second [19]. Stopped-flow
spectra, EPR and rapid quenching techniques were used for such
rapid reactions in order to understand their high rate in spite of the
spin prohibition [7]. The oxidized flavin (Flox) was monitored by its
strong absorption increase with a maximum at 450nm (Figure 1).
The short delay in the beginning indicated a reaction intermediate
formation. Addition of SOD inhibited the reaction; thus, the superoxide
involvement became clear at the first stage of the process
(Figure 2) [7].
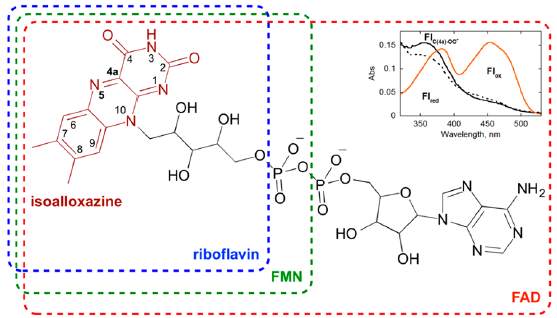
Figure 1: Structures of riboflavin (vitamin B2), FMN and FAD cofactors. Inset shows the UV−visible absorption spectra of the reduced (broken line) and oxidized flavin (red line), the C(4a)−peroxyflavin is presented by black line. From Ref. [18] with perimission.
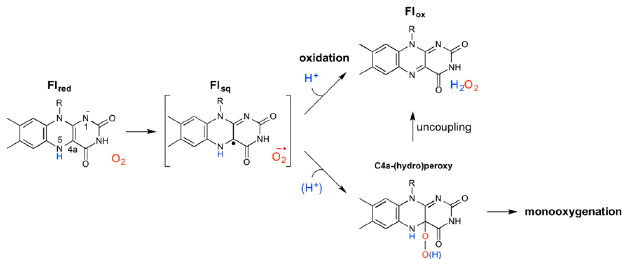
Figure 2: Typical dioxygen reactions catalized by oxidases and monooxygenases. Reactive atoms and positions are noted. From Ref. [18] with perimission.
This scheme was generalized later for flavoenzymes. The active center of enzyme is denoted in Figure 2 by square brackets. Electron transfer from reduced flavin (Flred) to O2 occurs as soon as dioxygen penetrates through protein channels into the active cite and occupies a close position to cofactor in the cavity between FAD and amino acid residues [1,2]. This result was supported first by density functional theory (DFT) calculations for the active center model of glucose oxidase [4]. Later on the similar mechanism was confirmed by numerous studies of a number of haem- and flavorenzymes, oxidoreductases, oxidases and oxygenases [22-44]. The radical pair between semiquinone (Flsq) and superoxide radicals (O2-•) in Figure 2 is originally generated in the triplet state because of the O2 triplet precursor. But the triplet state cannot produce products shown in Figure 2. From kinetic studies of reduced flavins (Flred) reactions with dioxygen in solvents Vincent Massey have proposed the following reaction scheme [19]:
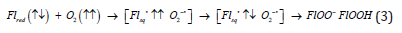
EPR spectra of semiquinones (Flsq) show a large spin density on C(4a) atom [7]. The product was identified as the C(4a)-flavin hydroperoxide (Figure 2) by detection of the 450 and 370nm absorption bands (Figure 1). In aqueous media this rather unstable product transforms heterolytically to the oxidized flavin (Flox) and H2O2 [19] (Figure 2). The late final product is typical for oxidase enzymes and intermediate flavinC(4a) hydroperoxide characterizes monooxygenases behavior (Figure 2) [23-51]. Thus, the mechanism of Eq. (3), proposed initially for reduced free flavins in solvents [19], was generalized for flavoenzymes (Figure 2) and postulates the following scenario. As in the solvent case, the electron transfer to O2 produces the caged triplet radical pair in the active cite of enzyme, which is capable (by unknown rea-sons) to undergo the T→S transition [18,19]. After such intersystem crossing, de-noted in Eq. (3), the normal singlet state chemistry follows and the singlet state radical pair proceeds to the short-lived FlOO- singlet species, which transforms into the intermediate C(4a)-flavin hydroperoxide (in monooxygenases) or into the hydrogen peroxide and oxidized flavin (Flox) in oxidases (Figure 2).
Massey [19] and all his ancestors [20] accept the spin flip at the radical pair stage in Eq. (3) for granted (without any comments or explanations). One can propose that Massey has conceived the arguments of the well-known radical pair theory (RPT) based on the cage effect in solvent and account for hyperfine coupling with nuclear spins. RTP has been proposed 50 years ago [33] being very popular in nineties [19]. But, the RTP ideas cannot be applied to enzyme active center, where the radical pair is not separated in two radicals by a long distance (> 10nm like in the solvent cage) [21]. Instead of very weak hyperfine interactions between electron and nuclear spins, which cannot induce T-S transition in flavoenzymes by no means, one has to consider another driving force to provide the spin flip in reaction presented in Eq. (3). If we suppose that only thermodynamic parameters (like in traditional chemistry) govern chemical reactivity of O2 molecule, rather than the spin-dependent kinetics, then in this case all organic part of the Earth’s biosphere would be consumed rapidly and converted into carbon dioxide and water; thus, aerobic life would be impossible. Life does exist because of specific internal magnetic interactions in the dioxygen open shell in a huge number of enzymes specified by Eq. (3).
Spin-Orbit Coupling in Dioxygen Open Shell
Now we have to consider O2 structure in more details. The
dioxygen molecule in the triplet ground state 3
O2 (X Σ−g ) is
characterized by the following electronic configuration (1σg)2
(1σu)2 (2σg)2 (2σu)2 (3σg)2 (1πu)4 (1πg)3 [9]. The first 14 electrons
occupy 7 low-lying molecular orbitals (MO) by pairs with opposite
spins. The 1πu and 1πg MO are doubly degenerate; thus, the two
outer electrons have to be dis-posed in two degenerate 1πg–MOs.
According to molecular analog of the Hund rule this implies to set
two non-paired electrons with the same spin orientations. Such
configuration provides the lowest triplet state of the type [ ↑ ][ ↑ ],
where the quantum cells denote two degenerate 1πg-orbitals. The
Hund rule and these two unpaired electrons in antibonding 1πg-
MOs are responsible for the specific character of dioxygen reactivity
in combustion and in bio-oxidation. Two antibonding 1πg-vacancies
make possible to transform dioxygen into reactive species like
superoxide O2-• and peroxide O2
2- anions. Next property of the
(1πg)2 open shell determines probability to generate the other
reactive species - singlet oxygen O2(a1Δg) [1]. One can construct
four possible quantum states, Eqs. (4) and (5), for the electronic
configuration (1πg)2. Let us consider cylindrical coordinate system,
where θ is an angle between the radius vector (r) and the molecular
axis (z), φ is the rotation angle of the radius vector about the z axis.
In this coordinate system the wave functions for two degenerate
πg–MO’s have a form |π±g>= ψ(r,θ) e±iφ. They are eigenfunctions
of the orbital angular momentum projection Lz on molecular z axis: 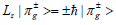 . Using this imaginary form of πg–MOs, the
quantum states of O2 and O2- molecule can be represented by the
following scheme [1]:
. Using this imaginary form of πg–MOs, the
quantum states of O2 and O2- molecule can be represented by the
following scheme [1]:
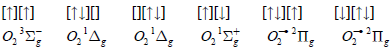
These configurations of the πg open shell are also eigenfunctions of Lz operator with eigenvalues 0, ±2ћ, 0. Exchange interaction stabilizes the triplet state |3Sg-> as the lowest one; the degenerate singlet states ∣1Δg> are higher in energy by 22kcal/mol, while the ∣1Σg+> state is the highest one with a total energy 36kcal/mol [9]. Wave functions of these states in the form of Slater determinants are:
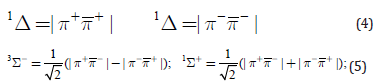
Some symbols are omitted here and only two-electronic parts of wave functions are shown [1]. Spin orbit coupling (SOC) between S and L angular momenta is known to be responsible for magnetic spitting of orbitally degenerate spin multiplets in atoms and diatomic molecules [9]. For diatomics such splitting is possible for states with the wave functions being eigenfunctions of the Lz operator (Lz Ψ= Λћ Ψ), for which Λ≠0 [9]. Naturally, magnetic spitting indused by SOC is possible only for states with spin quantum number S≠0 [9]. In such case the spin and orbital angular momenta projections on the molecular axis are observable values and their coupling depends on mutual orientation of both magnetic moments. This provides a well-seen SOC-induced splitting of the molecular Π, Δ, Φ multiplets (typically of the order 0.01eV in light molecules) [36]. In this work we use a simple semiempirical approximation for spin-orbit coupling (SOC) operator [10]
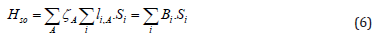
Here ζ А - is a spin-orbit coupling constant for the valence shell of atom A, deter-mined from atomic splitting, - are the orbital and spin angular momenta operators (in ћ units) for the i-th electron [1]. Expectation value of this operator is equal zero for the ground triplet state X3Σg- of the O2 molecule, since it has Λ=0. But the Sz =0 component of the triplet state, shown in Eq. (5), is still slightly split from the upper Sz = ±ћ components in the second order of perturbation theory, ΔE(2), since the triplet and singlet states in Eq. (5) are mixed by spin-orbit coupling matrix element [11]:
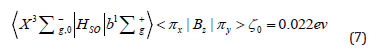
This integral is reduced to the spin-orbit coupling constant for the atomic oxygen O(3P) ground state and this is the largest possible energy of magnetic internal interaction in all oxygen all atrops. In the absence of external magnetic field the spitting between spin sublevels ms =0 and ms =±1 of the ground triplet state X3Σg- of the O2 molecule, produced by SOC contribution from Eq. (7) [10] is equal to:
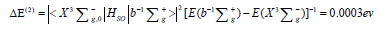
Here the S-T energy gap is equal 1.63 eV (from experiment [9]). The calculated ΔE(2) spitting is equivalent to 2.42cm-1, which provides 61% of the total observed zero- field splitting (ZFS=3.98 cm-1) [10]. The rest is determined by direct spin-spin coupling of two electrons [45-47]. This rather small measure of internal magnetic forces in the dioxygen ground triplet state have no direct connection with the origin of O2 activation by enzymes. However, it is important for the singlet dioxygen excitation and quenching in solvents [11-13]. Account of Eq. (7) in the first order of perturbation theory provides a mixture of two states from Eq. (5).
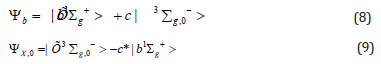
where coefficient is equal 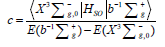 =0.0135. This small co-efficient of magnetic admixture between
ground triplet and excited singlet states is very important, being
responsible for many peculiarities of dioxygen [1]. It explains a
magnetic transition moment for X→b absorption and intensity of
the famous Fraunhofer line at 760nm in the Sun spectrum [15],
intensity borrowing scheme for a-X transition from the Noxon a-b
band in solvents and many others optical O2 effects [1]. Thus, the
SOC integral, Eq. (7), plays a great role in the theory of oxidative
stress and toxicity of reactive oxygen species [15]. The dioxygen
activation by flavoenzymes in our spin-magnetic treatment means
such mechanistic pathways that cause the release of spin prohibition
in O2 reactions with diamagnetic substrates, Eq. (1), making them
effectively allowed [2]. It is important that T and S wave functions
in Eq. (5) differ by electron rotation around z-axis; this creates
appearance of orbital angular momentum during the T-S transition,
which in turn simultaneously provides a torque to realize the spin
flip [22]. The similar quantum-mechanical ideas can be applied to
explain mysterious spin flip in Eq. (3) of Vincent Massey [2,22].
Let us consider electronic configuration of superoxide ion O2-• by
addition one more electron to dioxygen: (1σg)2 (1σu)2 (2σg)2 (2σu)2
(3σg)2 (1πu)4 (1πg)3 [2]. For such (1πg)3 open shell in the absence
of SOC account we have two degenerate configurations (a) and (b)
with ms=1/2, and similar two (a*,b*) - for ms=-1/2.
=0.0135. This small co-efficient of magnetic admixture between
ground triplet and excited singlet states is very important, being
responsible for many peculiarities of dioxygen [1]. It explains a
magnetic transition moment for X→b absorption and intensity of
the famous Fraunhofer line at 760nm in the Sun spectrum [15],
intensity borrowing scheme for a-X transition from the Noxon a-b
band in solvents and many others optical O2 effects [1]. Thus, the
SOC integral, Eq. (7), plays a great role in the theory of oxidative
stress and toxicity of reactive oxygen species [15]. The dioxygen
activation by flavoenzymes in our spin-magnetic treatment means
such mechanistic pathways that cause the release of spin prohibition
in O2 reactions with diamagnetic substrates, Eq. (1), making them
effectively allowed [2]. It is important that T and S wave functions
in Eq. (5) differ by electron rotation around z-axis; this creates
appearance of orbital angular momentum during the T-S transition,
which in turn simultaneously provides a torque to realize the spin
flip [22]. The similar quantum-mechanical ideas can be applied to
explain mysterious spin flip in Eq. (3) of Vincent Massey [2,22].
Let us consider electronic configuration of superoxide ion O2-• by
addition one more electron to dioxygen: (1σg)2 (1σu)2 (2σg)2 (2σu)2
(3σg)2 (1πu)4 (1πg)3 [2]. For such (1πg)3 open shell in the absence
of SOC account we have two degenerate configurations (a) and (b)
with ms=1/2, and similar two (a*,b*) - for ms=-1/2.
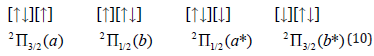
With account of spin-orbit coupling, Eq. (6), in the first order of perturbation theory these four configurations will be spit in two sublevels: E(2Π1/2) –E(2Π3/2) = ζ O [2]. This is in a good agreement with experimental result for O2-• ion: E(2Π1/2) –E(2Π3/2) = 160 cm-1 (0.02 eV) [48]. Such energy is relatively large for SOC in light molecule [49,50] and is determined by the observable orbital angular momentum of the 2Π state (Lz=ћ). It is million times larger than a typical hyperfine coupling used in the radical pair theory to induce T-S flip [33]. The same type of orbital rotation around z axis, as in Eq. (7), determines this internal magnetic energy. The splitting is inverted (Ω=3/2 is lower than Ω=1/2 sublevel), since the (1πg)3 open shell is more-than-half occupied [14]. Here Ω is the total angular momentum projection Ω=|Λ+ Sz| [9]. Now we can apply this result to the Massey’s mechanism of Eq. (3) [19], including our quantum cell notation [] for molecular orbitals and the curly brackets for the enzyme active center:
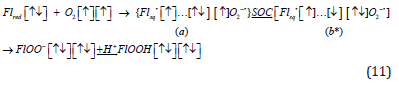
The triplet→singlet quantum transition in the enzyme active
center includes entirely the (a) → (b*) spin flip induced by SOC
inside the superoxide moiety [2,4]. The unpaired spin in flavin
semiquinone (Flsq) is inactive in the T-S transition, Eq. (11). Molecular
orbitals of the superoxide ion in the enzyme cage are perturbed
by protein scaffold and by cofactor; thus they are not completely
degenerate [4]. But molecular O2-• ion can rotate inside the cavity;
this hindered rotation will change the positions of the 2Π1/2 and 2Π3/2
sublevels and their splitting. Energies of all four configurations (10)
could be determined mostly by electrostatic perturbations rather
than by SOC [14]. This asymmetric top will change the sublevels
splitting and order with the frequency of libration movement. Thus,
the energies of the (a) and (b*) configurations would be close to
resonance periodically in the picoseconds time scale. The same is
true for the (b) and (a*) configurations in Eqs. (10)-(11). The most
important is the fact that spin-orbit coupling between such T and
S states is always large and close to 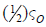 , since the structure
of two πg–MO’s are not changed much [1]. In superoxide ion
they are similar to πg,x and πg,y orbitals of O2 molecule and their
structure is not perturbed much in the enzyme active center. Small
admixtures from the nearest neighbors do not change the orbital
torque between (a) and (b*) configuration in the scheme, Eq. (11),
which remains similar to the orbital rotation in Eq. (7). The (½)
coefficient is the only main difference between SOC matrix element
in scheme of Eq. (11) and in Eq. (7). This factor (½) always enters
the T-S matrix elements of SOC operator, but Eq. (7) is a particular
exclusion because of specific structure of wave func-tions (5) [10].
, since the structure
of two πg–MO’s are not changed much [1]. In superoxide ion
they are similar to πg,x and πg,y orbitals of O2 molecule and their
structure is not perturbed much in the enzyme active center. Small
admixtures from the nearest neighbors do not change the orbital
torque between (a) and (b*) configuration in the scheme, Eq. (11),
which remains similar to the orbital rotation in Eq. (7). The (½)
coefficient is the only main difference between SOC matrix element
in scheme of Eq. (11) and in Eq. (7). This factor (½) always enters
the T-S matrix elements of SOC operator, but Eq. (7) is a particular
exclusion because of specific structure of wave func-tions (5) [10].
The T and S configurations of the (a) and (b*) types have been first taken into account for explanation of the charge-transfer contributions in SOC enhancement during the singlet oxygen quenching by aliphatic amines [49]. In the triplet oxygen activation by flavins and flavoenzymes presented in Eq. (11) this SOC effect is realized in the most simple and clear form [1-4,49]. The particular electronic structure of the superoxide ion, Eq. (10), provides very efficient magnetic torque from πg orbital rotation, which induces by SOC operator the spin flip during T-S transition. The twisting force that tends to cause spin rotation in the mechanism described by Eq. (11) is unusually strong for such light molecule as dioxygen. That is why the Massey’s mechanism is so efficient in many enzymes [22-30]. The T-S matrix element of SOC operator in enzyme active center described by Eq. (11) is equal to i(½)ζ O = i0.01 eV being imaginary value [2]. Calculation of the T→S transition rate constant includes the square of this absolute value [4]; thus, the imaginary unit (i=√-1) does not matter for the measurement of rate constant and can be omitted. The SOC energy of 10-2 eV realized inside small O2-• species (about 0.2nm diameter) corresponds to magnetic force of about 10-6 dyne. This is relatively strong force for quantum microparticles. We mean the force exerted between spin magnet and orbital movement (πg,x → πg,y rotation) of electrically charged particle (electron). Such force is much stronger than those magnetic forces, which deter-mine rate of T-S transitions in phosphorescence of organic molecules [50]. The rate constant for T-S transition in the caged radical pair of flavoenzyme shown in scheme of Eq. (11) can be in the range 104-106 s-1 [12,39]. Such fast intersystem crossing rate can compete with other possible relaxation rate processes at the elec-tron transfer stage in the enzyme active center, Eq. (11) [12-17,51]. Decay of the radical pair and the superoxide leakage from the cage of enzyme are slow processes hindered by the tight protein scafold (though neither flavin nor dioxygen provide covalent binding with nearest amino acid resudues) [5-7]. When the reduced oxidases react with dioxygen and form H2O2 these reactions have been investigated by stopped-flow techniques for a number of flavoenzymes, like GO, cholesterol, alditol and sarcosine oxidases, p-hydroxyphenyl acetate hydroxylase [7,19]. The measured rate constants demonstrate pseudo-first-order character being linear in dioxygen concentration with effective bimolecular rate constant of the order 106 -107 mole-1 s-1 [18,19]. These reactions are much faster than O2 reactions with free flavins reduced in solvents with rate constants about 250 mole-1 s-1 [5,18]. This means that protein environment of enzyme not only as-sists in the electron transfer to O2 [4], but also enhances vibrational maintenance of intersystem crossing process [39].
Cofactor-Independent Oxygenation
From the mechanism of Eq. (11) it is clear that flavin
cofactor does not contribute to the SOC matrix element 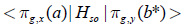 , since the nonpaired electron spin
of flavin semiquinone (Flsq) plays only role of a passive spectator
[17]. The role of Flavin cofactor is to provide electron transfer
for O2 eduction and to arrange proper binding of intermediates
in the course of reaction pathway on the singlet state potential
energy surface. The redox properties of Flavin are the main factor
of their enzyme activity. If the organic substrate possesses similar
redox potentials it can himself activate dioxygen for the spin flip
without cofactor assistance [17]. Such cofactor-free enzymes are
really discovered during last decade [34,35,52-54] and considered
as a new paradox of dioxygen activation [55]. In this respect, an
example of the bacterial 2,4-dioxygenase from Arthrobacter nitro
guajacolicus Ru61a, which operates without any cofactor and can efficiently degrade quinolones [52], attracts much theoretical
attention [17,22,27,31,34]. This 1-H-3-hydroxy-4-oxoquinaldine
2,4-dioxygenase (enzyme named as HOD) [33] is capable to
decompose 2-alkyl-3-hydroxy-4(1H)-quinolone (AHQ) substrate
by dioxygen and produce N-acetyl-anthranilate + CO products
[52]. On the ground of DFT calculations Hernández-Ortega, et al.
[31] have proposed a new mechanism based on primary triplet
diradical intermediate 3I1 produced by dioxygen covalent binding
with substrate at 2-position. The authors of Ref. [31] assumed
(without SOC calculations) that T-S transition occurs at the peroxyl
diradical 3I1 stage with subsequent intersystem crossing to the
singlet 1I1 peroxide inter-mediate followed by usual chemical
decomposition of quinolone to anthranilate at the 1I2 stage with
typical decarboxylation.
, since the nonpaired electron spin
of flavin semiquinone (Flsq) plays only role of a passive spectator
[17]. The role of Flavin cofactor is to provide electron transfer
for O2 eduction and to arrange proper binding of intermediates
in the course of reaction pathway on the singlet state potential
energy surface. The redox properties of Flavin are the main factor
of their enzyme activity. If the organic substrate possesses similar
redox potentials it can himself activate dioxygen for the spin flip
without cofactor assistance [17]. Such cofactor-free enzymes are
really discovered during last decade [34,35,52-54] and considered
as a new paradox of dioxygen activation [55]. In this respect, an
example of the bacterial 2,4-dioxygenase from Arthrobacter nitro
guajacolicus Ru61a, which operates without any cofactor and can efficiently degrade quinolones [52], attracts much theoretical
attention [17,22,27,31,34]. This 1-H-3-hydroxy-4-oxoquinaldine
2,4-dioxygenase (enzyme named as HOD) [33] is capable to
decompose 2-alkyl-3-hydroxy-4(1H)-quinolone (AHQ) substrate
by dioxygen and produce N-acetyl-anthranilate + CO products
[52]. On the ground of DFT calculations Hernández-Ortega, et al.
[31] have proposed a new mechanism based on primary triplet
diradical intermediate 3I1 produced by dioxygen covalent binding
with substrate at 2-position. The authors of Ref. [31] assumed
(without SOC calculations) that T-S transition occurs at the peroxyl
diradical 3I1 stage with subsequent intersystem crossing to the
singlet 1I1 peroxide inter-mediate followed by usual chemical
decomposition of quinolone to anthranilate at the 1I2 stage with
typical decarboxylation.
Other authors [17,22,39] have shown that SOC calculation
cannot approve such intersystem crossing mechanism since the
covalently bound 3I1 peroxyl diradical has no enough internal
magnetic force to induce efficient spin flip; orbital rotation is
quenched by O2 chemical binding at the 3I1 stage [22]. At the same
time the electron transfer and superoxide formation are possible
at the first stage of HOD enzyme catalysis [27]. Redox properties
of quinolone AHQ afford such electron jump from deprotonated
substrate anion to O2. In this case the efficient SOC in the triplet
radical pair between O2-• and deprotonated substrate radical
is emerged by the same mechanism as in the flavoenzymes
catalysis, Eq. (11), since such spin-orbit coupling is determined
entirely by the πg open shell of superoxide ion [22]. The spin flip
(T-S transition) induced by efficient SOC does happen and the
simple chemical transformations proceed further on the singlet
state potential energy surfaces according to calculation scheme
presented in Ref. [31]. The main feature of this catalysis by HOD
and by other cofactor-free enzymes [55] is that their substrates
are capable for deprotonation; thus, substrate anion himself can
catalyze an electron transfer to O2 and generate substrate radical
and superoxide ion radical pair in the initial triplet state. Then our
mechanism of internal magnetic interaction inside O2-• moiety,
shown in Eq. (11), can be applied for all cofactor-free enzymes of
similar type [35,55]. Internal magnetic property of the superoxide
open shell, Eq. (10), provides orbital torque, which is responsible for
T-S transition in such radical pair [2]. After this spin flip the singlet
state with paired spins can recombine to form a hydroperoxide
intermediate followed by other singlet state reactions toward
selective products [34,35]. The main driving force for all considered
oxidation and oxygenation processes is a magnetic torque created
by the (a)→(b*) electron jump with simultaneous spin flip, shown
in Eq. (11). Dioxygen activation by all considered enzymes [22-
44,51-56] starts with the triplet radical pair formation which
includes superoxide ion. Spin-orbit coupling in the O2-• moiety, Eq.
(10), provides effective removal of spin prohibition through the
high rate of intersystem crossing (T-S transition) in such radical
pair [1-4]. All biochemists must be convinced in this mechanism of
O2 activation since the strength of magnetic force responsible for
the spin flip is proved by experimental measurement of the spin
splitting in the ground doublet state X2Πg of superoxide ion [48].
Conclusion
Oxidation of organic matter by the triplet O2 molecule from the
air (dioxygen) is a spin-forbidden process in the absence of ignition
by radicals. Reactivity of dioxygen starts to attract a new interest
recently by few reasons. This is a challenge for basic researches,
which aim to highlight the most fundamental aspects of dioxygen
chemistry being not solved since the Lavoisier and Faraday
discoveries. The former has shown that O2 gas is responsible for
respiration and combustion; the late - determined para magnetism
of dioxygen. It became clear now that only modern quantum
chemistry at the level of relativistic theory can approach to solution
of the metabolic oxygenation and respiration problems. In
2002 the author of this review has proposed a general answer
to the question “How enzymes can activate the triplet ground
state dioxygen for organic substrates oxidation?” [2] The answer
reduces this complicated biological problem to analysis of weak internal
magnetic perturbations in the active sites of two enzymes,
glucose oxidase and copper amine oxidase [2]. A number of recent
publications support these ideas and extend their applications to
various enzymes. One can see that reduced flavin in the singlet
ground state initially reacts with the triplet dioxygen by a single
electron transfer, followed by the rapid T-S transition with
subsequent formation and decay of a covalently bound intermediate.
A common characteristic of the oxygen-utilizing flavoenzymes is the
dramatic rate increase in comparison with free flavins for the triplet
dioxygen reduction. Some monooxygenases are able to produce
flavin hydroperoxide as a metastable intermediate and then to use
this reactive species in various useful synthesis as nucleophile or
electrophile reagents. With new knowledge of magnetic physical
mechanisms of dioxygen activation these could be useful synthetic
targets for drug design with high medical selectivity and special
purposes.
All known flavoenzymes able to catalyze the triplet O2 reactions
with organic substrates not only accelerate the rates of those
reaction, but also govern the reactions along particular reaction
paths, which lead to selective products [49-54]. This is determined
by fast electron transfer and by T-S spin flip assisted by special
organization of the nearest protein environment. DFT calculations
show that some amino acid residues assist to provide electron
transfer to dioxygen and influence spin-orbit coupling effect on
T→S transition in the enzyme active center. This was shown first for
oxidation half reaction in glucose oxidase from Aspergillus Niger [4]
and latter supported by DFT calculations for many other enzymes [22-44].
The rate of spin flip strongly depends on protein scaffold
structure as well as the subsequent chemical reactions along the
singlet pathways.
Advent of photosynthesis and Oxygen Catastrophe in the first
half of the Earth’s billion-years history shacked the world and
life evolution in such a way that eukaryotes got accustomed to
cope with O2 in the air and developed various enzymes including
superoxide dismutase [56]. Metal containing enzymes are able to
overcome spin prohibition for O2 reactions with organic matter by
simple spin-catalysis based on exchange interaction. The late has
a natural origin being typical for chemical bonding, which, by the
way, explains an easy oxidation of metals by air. The most difficult
(from the mechanistic point of view) and unusual for traditional
chemistry laws is a wide type of enzymes, which do not contain
metals. The crucial chemical role in these enzymes plays a weak
magnetic force in-side the superoxide πg open shell, Eq. (10), which
induces the T→S quantum transition and abolishes the severe spin
prohibition on dioxygen reactivity. The unfavorable kinetic barrier
for organic oxidation by the triplet O2 molecule is relaxed in the
enzyme active center by one-electron reduction to O2-•, which bears
inside it-self a crucial magnetic force to induce the necessary spin
flip leading to selective products without damage for the cellular
milleu. One can say that this is the main driving force of all aerobic
evolution on the Earth.
For more
Articles on : https://biomedres01.blogspot.com/



No comments:
Post a Comment
Note: Only a member of this blog may post a comment.