The Diagnostic Difficulties and The Delayed Identification of Anaplastic Large Cell Lymphoma in A Teenage Patient. A Review of Literature
Introduction
Anaplastic large cell lymphoma (ALCL) originates from peripheral T-lymphocytes and is the most common mature T-cell neoplasm in children and adolescents. All in all, these neoplasms are relatively rare among all types of lymphomas. ALCL comprises about 10-20% of all non-Hodgkin lymphomas (NHL) in the above group of patients [1-3]. Essentially, this lymphoma does not occur frequently and is often difficult to distinguish, though its identification is essential for both the prognosis and treatment [4]. The primary site of the lymphoma can appear in almost any location, including lymph nodes. This may result in unspecific clinical signs of disease leading to why it can take several months from the appearance of the first symptoms to make an accurate diagnosis.
ALCL was recently classified by the World Health Organization (WHO) as primary cutaneous ALCL (C-ALCL) and systemic ALCL (S-ALCL). The second category was further divided into two distinct types based on expression of a protein called anaplastic lymphoma kinase (ALK), ALCL ALK-positive (ALK+) and ALCL ALKnegative (ALK-) [5]. The presence of this protein on neoplastic cells is more frequent in children and young adults, with a median age of approximately 30 years, whereas the lack of it is proven in older adults (median age of about 55 years). Simultaneously, this feature is associated with improved overall survival [2,6,7]. Additionally, the assessment of ALK expression plays an essential role in the case of S-ALCL. The overall survival rate of ALK+ lymphoma increases up to 70% and is far better that of ALK- lymphoma that ranges from 15-45% [6,7]. C-ALCL is marked by indolent development and a favorable prognosis with a survival rate of 80-95% [2]. We present the case of a patient with an unusual clinical manifestation and difficulties in the diagnostics of S-ALCL. We would like to point out what to do in order to avoid mistakes in the early stages of this disease. Additionally, attention should be paid to the aggressive and insidious growth of S-ALCL in a very brief time.
Case Presentation
Patient History and Information
A sixteen-year-old boy was admitted to the Surgery Clinic in July 2017 due to a nodular skin lesion. The 4 cm lesion was located in his lower abdomen and was darker than the surrounding skin. On clinical examination, the lesion was suspected to be an abscess and the patient was sent to surgery. The lesion was incised and did not contain any liquid content. No tissue material was taken for histopathological examination. The wound did not heal, was suppurating and slowly reducing its size. Furthermore, in August 2017, hard and painless tumors appeared in the area of both groins. Ultrasound (US) and computed tomography (CT) of the abdominal cavity and pelvis showed enlarged lymph nodes in the inguinal and pelvic regions. Based on the above examination, lymphoma was suspected, and the boy was sent to the Department of Pediatric Oncology, Hematology and Transplantology at Poznan University of Medical Sciences.
Diagnostic Assessment
On the day of admission in September 2017, the physical examination showed an unhealed wound on the skin of the lower abdomen and palpable hard nodal masses in the iliac fossae. We have only a photographic evidence of the scar 4 months after surgical treatment (Figure 1). Additionally, a painless fistula with leakage was observed inside the oral cavity in the area of the right molar teeth. The liver and spleen were not enlarged. The results of biochemical tests, morphology, and peripheral blood smears were within normal limits. During his stay in the Department, the patient presented with a low-grade fever and increasing facial asymmetry caused by swelling of the right cheek. CT scan and magnetic resonance imaging (MRI) of the head revealed the solid mass (60x42x35 mm) filled almost all of the space of the right maxillary sinus and destroyed the structure of the posterior wall of the sinus, invading the pterygopalatine fossa and adipose tissue, reaching the area of the masseter muscles. Laterally, it was spreading into the right buccal area where from the medial side, it was infiltrating the lateral part of the palate. Upwards, it was penetrating the inferior orbital fissure and damaging the upper wall of the maxillary sinus into the right orbit. There were no signs of the tumor spreading to the middle cranial fossa. Furthermore, numerous enlarged lymph nodes of the upper neck region were found. Biopsies of the inguinal node, exophytic craniofacial tumor, and bone marrow along were performed. The patient was also examined with positron emission tomography (PET), which allowed for a determination of disease in stage IV.
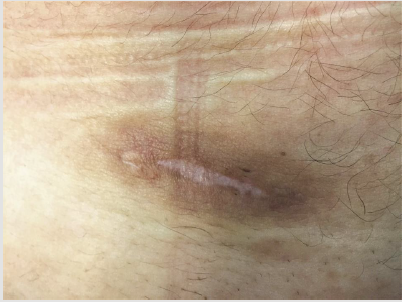
Figure 1: The scar in the lower abdomen 4 months after surgical treatment of the primary lesion.
Histopatological Findings
Materials and Methods: The specimen was taken from three different sites and sent to Pathology Department. The first surgery consisted of excisional biopsy of enlarged, inguinal lymph node. 5 days later the biopsy of bone marrow (two trephines) was done. 6 days later excisional biopsy of two tumors in the oral cavity, (including right maxilla and oral vestibule) was performed. All resected specimens were fixed in 10 % buffered formalin, routinely processed and embedded in paraffin, stained with hematoxylin and eosin. An initial panel of immunohistochemical stains was performed on an automated immunostainer (Ventana Benchmark, Roche/Ventana Medical Systems, Tucson, AZ) and this included: CD30, EMA (Epithelial Membrane Antigen), ALK-1, CD43, CD3, CD15, CD20, Desmina (DES), CD25, Granzyme B, Perforin, TIA 1, CD7, CD5, Cd8, Cd56, MUM 1, PDL 1, CD4, CD2, EBI-1, PAX-5, Ki 67.
Results: Grossly, the first resected specimen consisted of
solid
soft tissue in form of a lymph node measuring 3,5 cm x 3,0 cm x
2,0 cm. It was oval in shape, but the external surface was rather
smooth. On cross section, the tumor was white to grey, solid
and encapsulated. The second specimen consisted of two core
biopsies specimen of bone marrow measuring 2,6 cm and 0,8 cm
in length. The third specimen from the oral cavity consisted of two
separated specimens each of which was fragmented measuring 5
cm and 3 cm respectively in greatest diameter. Microscopically, the
neoplasm invaded the lymph node and the specimens from oral
cavity. The bone marrow specimen was not impaired. The tumor
cell occupied almost entire parenchyma of the lymph node (Figure 2a).
The neoplastic cells differed in size from small to large ones
(Figure 2b). This was helpful in distinguishing ALCL from Hodgkin
Lymphoma. The cells invading the lymph node and the oral cavity
had the same morphology (Figure 3). Most of the neoplastic cells
were large, irregular and bizarre. They often had polylobated nuclei
and the subset of cells had eccentric horse- or kidney-shaped nuclei
consisted with Hallmark cells characteristic of ALCL. Admixed with
the tumor were scattered small lymphocytic cells. Mitotic figures
were easily encountered (60 per 10 high power fields). There was
no necrosis.
Immunohistochemically the tumor cells stained for CD 30 and
ALK 1 showing nuclear and cytoplasmic staining (Figure 4a and
4b). Such pattern of staining correlates with underlying genetic
abnormality t (2;5). They also stained for EMA, CD43, CD25,
Granzyme B, Perforin, TIA 1, Ki -67 in 80% of the cells and focally
for CD3, CD7, CD5, CD8, CD56, MUM 1, PDL 1. They neoplastic cells
were negative for CD15, CD20, Desmin, CD4, CD2, EBI-3, PAX 5.
The microscopic and immunophenotypic studies confirmed the
diagnosis of Anaplastic Large Cell Lymphoma, common type.
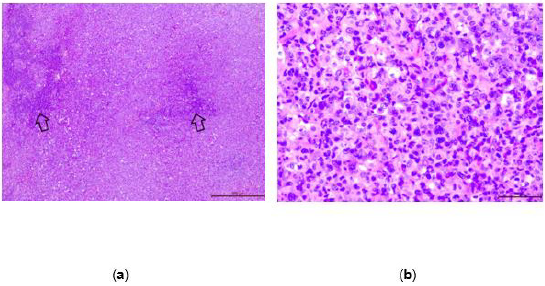
Figure 2: (a) The tumor invades almost entire inguinal lymph node tissue. There are only small areas of the normal lymph node (arrows), H&E, 200X; (b) The neoplastic cells range from small to large ones. They usually have large, irregular, and bizarre or polylobated nuclei, H&E ,400X.
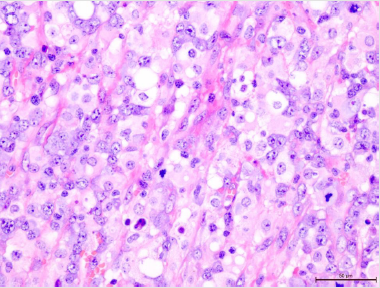
Figure 3: The neoplastic cells infiltrating the mucosa of the oral cavity, H&E, 400X.
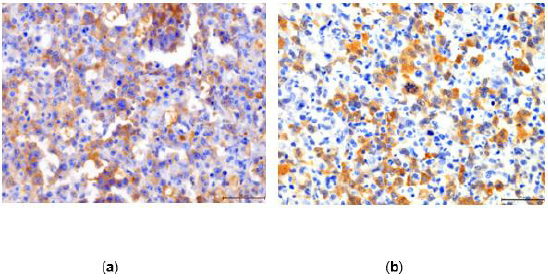
Figure 4: (a) The tumor cells are diffusely positive for CD30, 400X; (b) Most of the neoplastic cells stain for ALK 1, 400X.
Diagnosis
Based on the PET scan and pathology report, ALCL in stage IV was diagnosed.
Treatment and Outcome
After establishing the final diagnosis, treatment according to the international protocol ALCL 99 was started. It consisted of six alternating, consecutive courses of AM (dexamethasone, methotrexate, ifosfamide, cytarabine, etoposide) and BM (dexamethasone, methotrexate, cyclophosphamide, doxorubicin) [8]. Currently, the patient remains in the first complete remission of the disease, 36 months after their treatment.
Discussion
S-ALCL remains a moderately aggressive lymphoproliferative
neoplasm with worse outcomes than most B-cell lymphomas, yet
significantly better survival than other peripheral T-cell lymphomas
[9]. The majority of patients are male and present with advanced
disease in stages III or IV, often with systemic symptoms, especially
fever. Dissimilar from adults, ALCL in children usually demonstrates
expression of ALK [1,3,6,7]. The diagnosis of S-ALCL can be
challenging, mainly when first signs appear in the skin. One of the
reasons for this is that both types of S-ALCL can secondarily involve
the skin and C-ALCL can secondarily involve regional lymph nodes.
It shows the importance of the correlation of histopathological
findings with staging and other clinical data in order to establish
a proper diagnosis [9]. The stage of the disease at the beginning is
frequently determined as advanced. It may often result from both
a late diagnosis as well as a rather rapid developmental rate of
disseminated neoplastic cells. In literature, such cases take a few
months from the first signs of disease (Table 1) [10-15]. In our case,
that period lasted three months.
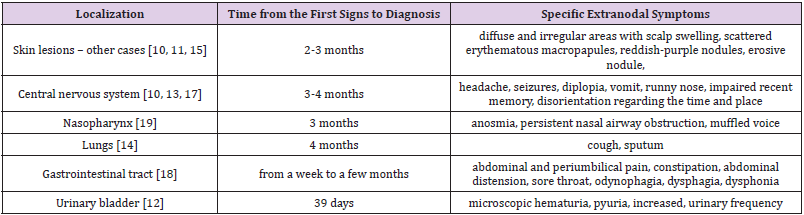
Table 1: The review of the approximate time from the first presenting signs to diagnosis and distinctive symptoms resulting from particular extranodal sites of ALCL in the literature.
The primary site of S-ALCL can occur in almost any location
in the human body. In most cases at first, it affects lymph nodes,
and the initial sign is painless lymphadenopathy. If that happens,
the proper procedure is to carry out a broad differential diagnosis.
We should take into account bacterial and viral infections, zoonotic
diseases, connective tissue diseases and neoplasms, notably
leukemia and lymphomas. In 60% of ALK+ anaplastic large cell
lymphomas, there is an additional involvement of extranodal
tissues most often concerning skin, bones, bone marrow, soft
tissues, lungs and liver [1,6,7]. However, S-ALCL presenting initially
as a localized skin lesion is an infrequent finding, as was the case
with our patient [11,15]. His first sign of lymphoma was a nodular skin
lesion that resembled an abscess. Other primary extranodal
localizations of S-ALCL are presented in literature, which include
soft tissues, lungs, bones, stomach, small intestine, tonsils, and the
central nervous system [13,16-18].
Symptoms resulting from extranodal involvement (listed in
Table 1) may imitate various other diseases and linking them to a
lymphoproliferative neoplasm can be quite challenging. Examples
of an atypical clinical course of ALCL can be found in literature
[10,11,13,14,19]. In cases of involved lungs, the lesion may present
as tuberous masses, atelectasis, displacement of the mediastinum
to one side and pleural effusion [14]. Sites in the central nervous
system are accompanied by various neurological disorders such
as diplopia, nausea, vomiting, neck stiffness, persistent headaches,
impairment of recent memory, disorientation regarding the time and
place and poor state of mind [10,13]. Skin lesions can occur either
as a soft and swollen nodular lesion or as a scattered, erythematous
and maculopapular one, both of which can display ulceration [10,
11]. Moreover, a case of incorrect diagnosis of mononucleosis has
been reported in a teenage girl with a nasopharyngeal mass and
cervical lymphadenopathy where the mass caused nasal airway
obstruction [19].
Due to a wide range of complaints, patients may visit many
specialists in the early stages of disease. These physicians, who
are usually not oncologists, should keep oncological alertness in
every situation including trivial ones, as they play a particularly
important role in the initiation of the diagnostic process and referral
of patients to oncologists. The proper proceedings have a decisive
influence on prognosis, which also includes surgical interventions
of precarious lesions. For the described patient, a surgeon made
a significant mistake at the beginning of the diagnostic process.
Without any examination and histopathological analysis, the
doctor considered a nodular skin lesion as an abscess and incised
those changes to drain it. Thus, the time to make a diagnosis of
ALCL and initiating the treatment in the presented patient was
markedly protracted. The absence of any liquid content inside the
skin lesion should have led the physician to verify the previous
assessment. Moreover, he ought to have collected some material for
histopathological examination. What is more, the increased healing
time of this wound should have awaken the doctors’ suspicion of
the neoplastic character of this lesion.
Its histopathological features cause further difficulties
in diagnosing ALCL. The marked cellular pleomorphism and
morphologic variation of ALCL raises a broad differential
diagnosis including cancer, melanoma, sarcoma and other
hematopoietic neoplasms such as diffuse large B-cell lymphoma,
classical Hodgkin lymphoma, myeloid sarcoma and peripheral
T-cell lymphoma not otherwise specified [20]. ALCL shows a
broad morphological spectrum. Still, all cases contain a variable
percentage of cells with eccentric, horseshoe- or kidney-shaped
nuclei, often with an eosinophilic region near the nucleus, socalled
hallmark cells [3]. It is essential to recognize that rare cases
may have subtle morphological features, including a prominent
reactive background [20]. ALK+ and ALK- types of S-ALCL
share similar morphological features and diagnosis cannot be
established based on morphological evaluation alone. Thus, the
immunohistochemistry and molecular profile (IHC) play essential
roles in the diagnosis and subclassification of ALCL [9]. The ALK+
form is associated with the NPM-ALK t (2;5) translocation, which
highly correlates with the identification of the ALK protein by IHC
[2]. CD30 antigen expression in the neoplastic cells is distinctive of
ALCL, but it is not a specific marker for this lymphoma. However,
the strong and homogenous staining helps distinguish ALCL from
other CD30+ lymphomas [6,9]. Besides being CD30+, most cases
also express epithelial membrane antigen (EMA). Moreover, ALCLs
are considered peripheral T-cell lymphomas as they express one
or more T-cell antigens. Therefore, markers like CD2, CD4 and CD5
are positive in a significant proportion of cases. However, due to
the loss of several pan T-cell antigens, some cases may have an
apparent “null cell” phenotype [6,21]. S-ALCL is not one disease
entity, and the “one size fits all” treatment strategy is not ideal [8].
To implement the best targeted therapy, there is a necessity of an
exact determination of the neoplastic cells’ immunophenotype. In
view of the above, and due to additionally significant complexity
of histopathological diagnostics of lymphomas, this process should
be performed in the department of pathology with the highest
reference level. The facility should have all available diagnostic
techniques, beyond standard pathomorphological evaluation,
including but not limited to, broad antibody panels to IHC testing,
flow cytometry, cytogenetics and molecular diagnostics [22].
S-ALCL, especially in ALK+ peculiarities, exhibits chemosensitivity
in the front line and at relapse, leading to high response
rates with vastly different chemotherapy regimens [9,21]. The
beginning of the treatment based on an incorrect diagnosis may
cause the implementation of an incorrect chemotherapy protocol.
On the other hand, leaving the patient without any treatment
when the disease progresses very quickly can eliminate further
therapeutic options and lead to disastrous complications [22].
Results of the Savage et al. study showed that an advanced stage
and poor performance status are poor prognostic factors in both
ALK+ and ALK- ALCL. In contrast, multiple extranodal sites of
involvement and increased age are bad prognostic factors only in
the ALK+ type [6]. Implementation of targeted therapy at early
stages of disease may result in a better chance of recovery.
Conclusion
Anaplastic large cell lymphomas represent complicated diagnostic difficulties. The possibility of localization of both primary and secondary sites in extranodal locations makes clinical manifestation non-specific. Presented symptoms may seem trivial or can indicate another condition and as a result, can be easily marginalized. For this reason, it can lead to making a significant mistake at the beginning of the diagnostic process. Complicated pathomorphological diagnostics are based on immunophenotype evaluation with confirmation in the reference facility. As a consequence, ALCL is most often diagnosed in an advanced stage. Physicians who encounter such patients at the beginning are usually not oncologists. However, they are crucial to initiate the relevant diagnostic processes and refer the patient to the oncology center afterward. It is therefore essential to stay alert all of the time. Whether the patient will survive, improve and recover is their responsibility.
For more Articles on : https://biomedres01.blogspot.com/



No comments:
Post a Comment
Note: Only a member of this blog may post a comment.