Usher Syndrome, Retinosis Pigmentosa and Deafness, Case Report
Case Report
Here we present the case of a 22-year-old masculine patient,
referred by the Genetics Department, due to hearing disorders, to
rule out injuries that might compromise his sight. Medical history:
five-year evolution of progressive hearing loss and tinnitus. Rest:
denied. Non-pathologic personal background: denied. Maternal
uncle with visual and hearing deficiency without precise diagnosis.
Ophthalmological examination: visual acuity 20/40 right eye and
20/40 left eye. Eyelids and annexes were normal, transparent
anterior segment, cornea and lenses. Intraocular pressure 12
mmHg in both eyes. After fundoscopic examination, both eyes
present optic nerve slightly pale, diffuse retinal pigment changes
that show choroid vessels. Bottom in bilateral salt and pepper as
well as bone spicules (Figure 1). Right eye refraction +1.00, left
eye +0.50 spherical. An electroretinogram reports that there is no
recordable response in low scotopic stages. In photopic stages there
is a slight response of cones and flicker of low extent and abnormal
implicit time (Figures 2 & 3). As part of the protocol, an audiogram
was requested (Figure 4). It describes a downward profile curve
with medium deafness by bilateral sensorial pure tone (PT)
prevalence. Concluding, as a diagnosis, bilateral medium deafness
with neurosensorial prevalence. Campimetry presents advanced
24-2 + with suppression of total right eye deviation affecting
temporal hemifield and superior Bjerrum scotoma, respecting the
30 degrees of the nose. Left eye, temporal paracentral affectation
(Figure 5). Optical coherence tomography (OCT) of the optic nerve
was carried out, which showed thinning of nervous fibers in superonasal
quadrant with borderline values and superior and inferior
thinning in left eye. Macular OCT is requested, where affectation of
pigmentary epithelium of the retina is shown in an extensive way
(Figure 6).
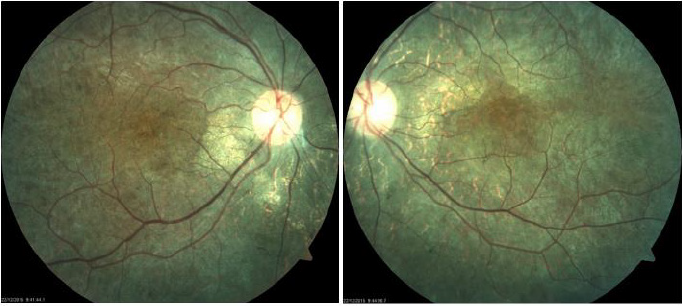
Figure 1: Fundoscopic findings: both eyes with “salt and pepper” changes, bone spicules, slight vascular thinning, optic nerve with “waxed” appearance.
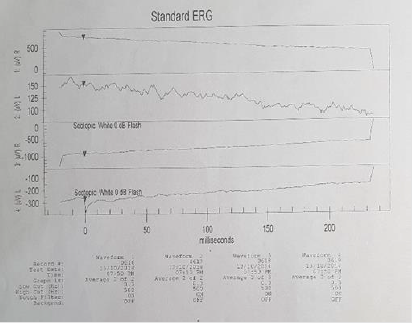
Figure 2: Response absence in scotopic conditions.
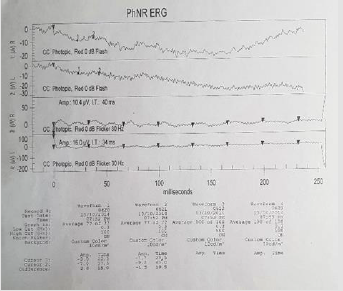
Figure 3: Slight response of cones, with low extent flicker.
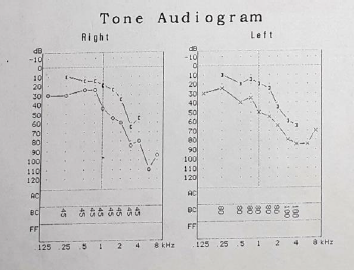
Figure 4: Moderate right deafness of a mixed type descending profile, Drop in treble. Moderate left deafness of a mixed type with descendent curve.

Figure 5: Visual fields. Right eye affectation in hemfield and scotoma superior Bjerrum. Left eye important temporal and nose paracentral affectation.
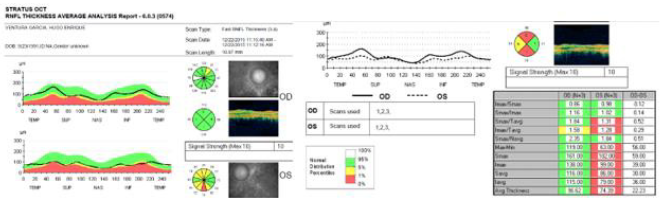
Figure 6: OCT (optic coherence tomography) of optic nerve, showing higher affectation and thinning in layer of nervous fibers left eye, in accordance with campimetric damage.
Discussion
Usher syndrome is traditionally subdivided in three types, depending on its clinic presentation; these are differentiated by the severity and progression of the hearing loss and the presence or absence of vestibular symptoms; the visual affectation is present in all of them [4]. Type 1 presents congenital profound sensorineural deafness, vestibular dysfunction, occurrence before puberty and progressive retinosis pigmentosa, whereby it is the most severe form of this syndrome and represents 30-40% of all cases. Five genes have molecularly been identified, which affect the VIIa myosin driver protein (the most common), in this type there is more probability of retina damage. Foveal atrophic lesions were identified only in patients with type 1 Usher syndrome and were of older age [4,5]. Type 2 is less severe than type 1 and is characterized by congenital moderate-severe deafness. The age of onset of the retinosis is variable [4]. Type 3 corresponds to patients that show progressive hearing loss, variable beginning of retinosis pigmentosa and vestibular disfunction with wider variability range. This type is the least common with a prevalence of 2-4% [4].In accordance with this classification, our patient belongs to type 3 depending on the clinic apparition of the hearing loss noted by the patient 5 years ago, while the ophthalmologic exploration study was recently done.
This syndrome presents a high clinic and genetic diversity. It can involve up to 49 genes. Some of them are identified in nonsyndromic deafness [4,6].
These mutations can be detected up to a 33%, although only one allele can be involved [4]. As genetic orientation, the patient has a family medical history of a maternal uncle with deafness and visual loss without a confirmed diagnosis. The retinosis pigmentosa is the pivot clinic finding to differentiate an Usher syndrome from a non-sensorial syndromic deafness. There is a degeneration of the organ of Corti and atrophy of the vascular groove. The first symptom is the night blindness due to the progressive degeneration of the rods but can get worse around the third or fourth decade [1]. There are reported cases of proliferative vase tumors. Even though the association between a retinal tumor of this type, with type 1 Usher syndrome is extremely rare [7]. This finding was not encountered in our patient. Other clinical findings include olfactory loss, abnormalities of nasal cilia, diminished spermatic mobility, general reduction of the brain mass, not only from the posterior nostril or visual and hearing systems. Several psychiatric disorders have been associated with Usher syndrome, as schizophrenia, atypical psychosis and frequent depression [1,3]. Other conditions with hearing loss and retinosis pigmentosa are the Waardenburg syndrome, Alport syndrome and Refsum sickness [7]. However, the severity of the clinical picture of these diseases and the age of the onset of symptoms dismiss any relation with our patient.
The visual exploration must include best corrected visual acuity, bio microscopy in slit lamp and ophthalmoscopy. Moreover, complimentary studies include campimetry, macular OCT, ocular fundus pictures, electroretinogram (ERG) [8]. The electroretinogram (ERG) is reported as the ideal study to find retinosis pigmentosa in early stages, and the guide of the Sociedad Nacional de Niños con Sordera recommends that children with sensorineural deafness must undergo this study even if there are no symptoms or signs. Some investigators have proposed, as a routine procedure, to make an ERG in all children with this type of deafness, but it is still under discussion the appropriate age to initiate the screening, which goes from 8 years old and on, or up to before 7 years old in case of congenital deafness. It must be considered that a normal ERG does not exclude the Usher syndrome diagnosis.9,5 Our case presents clear visual affectation in scotopic conditions, and delay in response and intensity to the cones in photopic phase. The prevalence of retinosis pigmentosa in revised patients is 15%, whereas in previous studies without this diagnosis method only reveal a 7 to 10.6%. Changes in the ERG occur even before the clinic manifestation or the signs to the fundoscopy [9]. The genetic study of Usher syndrome allows us to adequately evaluate the use of cochlear implant, genetic council and prenatal diagnosis [10]. Although an earlier study does not represent an extreme change in the treatment or prediction for the affected patients, it can help a genetic council with which the affected family can make decisions, and parents can search for a prenatal diagnosis for future children.
Conclusion
Usher syndrome is a condition that requires a multidisciplinary
attention from the first suspicion. Routine hearing and visual
screening should be made in pediatric patients, since the variability
in the presentation of this syndrome, and the genetic complexity,
important data to conclude its diagnosis at an early age could
escape. A complete study of this disease leads to new adaptation
opportunities of the hearing treatment as well as an adequate
genetic advice for parents and other family members.
Case Report
Here we present the case of a 22-year-old masculine patient, referred by the Genetics Department, due to hearing disorders, to rule out injuries that might compromise his sight. Medical history: five-year evolution of progressive hearing loss and tinnitus. Rest: denied. Non-pathologic personal background: denied. Maternal uncle with visual and hearing deficiency without precise diagnosis. Ophthalmological examination: visual acuity 20/40 right eye and 20/40 left eye. Eyelids and annexes were normal, transparent anterior segment, cornea and lenses. Intraocular pressure 12 mmHg in both eyes. After fundoscopic examination, both eyes present optic nerve slightly pale, diffuse retinal pigment changes that show choroid vessels. Bottom in bilateral salt and pepper as well as bone spicules (Figure 1). Right eye refraction +1.00, left eye +0.50 spherical. An electroretinogram reports that there is no recordable response in low scotopic stages. In photopic stages there is a slight response of cones and flicker of low extent and abnormal implicit time (Figures 2 & 3). As part of the protocol, an audiogram was requested (Figure 4). It describes a downward profile curve with medium deafness by bilateral sensorial pure tone (PT) prevalence. Concluding, as a diagnosis, bilateral medium deafness with neurosensorial prevalence. Campimetry presents advanced 24-2 + with suppression of total right eye deviation affecting temporal hemifield and superior Bjerrum scotoma, respecting the 30 degrees of the nose. Left eye, temporal paracentral affectation (Figure 5). Optical coherence tomography (OCT) of the optic nerve was carried out, which showed thinning of nervous fibers in superonasal quadrant with borderline values and superior and inferior thinning in left eye. Macular OCT is requested, where affectation of pigmentary epithelium of the retina is shown in an extensive way (Figure 6).
Figure 1: Fundoscopic findings: both eyes with “salt and pepper” changes, bone spicules, slight vascular thinning, optic nerve with “waxed” appearance.
Figure 2: Response absence in scotopic conditions.
Figure 3: Slight response of cones, with low extent flicker.
Figure 4: Moderate right deafness of a mixed type descending profile, Drop in treble. Moderate left deafness of a mixed type with descendent curve.
Figure 5: Visual fields. Right eye affectation in hemfield and scotoma superior Bjerrum. Left eye important temporal and nose paracentral affectation.
Figure 6: OCT (optic coherence tomography) of optic nerve, showing higher affectation and thinning in layer of nervous fibers left eye, in accordance with campimetric damage.
Discussion
Usher syndrome is traditionally subdivided in three types, depending on its clinic presentation; these are differentiated by the severity and progression of the hearing loss and the presence or absence of vestibular symptoms; the visual affectation is present in all of them [4]. Type 1 presents congenital profound sensorineural deafness, vestibular dysfunction, occurrence before puberty and progressive retinosis pigmentosa, whereby it is the most severe form of this syndrome and represents 30-40% of all cases. Five genes have molecularly been identified, which affect the VIIa myosin driver protein (the most common), in this type there is more probability of retina damage. Foveal atrophic lesions were identified only in patients with type 1 Usher syndrome and were of older age [4,5]. Type 2 is less severe than type 1 and is characterized by congenital moderate-severe deafness. The age of onset of the retinosis is variable [4]. Type 3 corresponds to patients that show progressive hearing loss, variable beginning of retinosis pigmentosa and vestibular disfunction with wider variability range. This type is the least common with a prevalence of 2-4% [4].In accordance with this classification, our patient belongs to type 3 depending on the clinic apparition of the hearing loss noted by the patient 5 years ago, while the ophthalmologic exploration study was recently done.
This syndrome presents a high clinic and genetic diversity. It can involve up to 49 genes. Some of them are identified in nonsyndromic deafness [4,6].
These mutations can be detected up to a 33%, although only one allele can be involved [4]. As genetic orientation, the patient has a family medical history of a maternal uncle with deafness and visual loss without a confirmed diagnosis. The retinosis pigmentosa is the pivot clinic finding to differentiate an Usher syndrome from a non-sensorial syndromic deafness. There is a degeneration of the organ of Corti and atrophy of the vascular groove. The first symptom is the night blindness due to the progressive degeneration of the rods but can get worse around the third or fourth decade [1]. There are reported cases of proliferative vase tumors. Even though the association between a retinal tumor of this type, with type 1 Usher syndrome is extremely rare [7]. This finding was not encountered in our patient. Other clinical findings include olfactory loss, abnormalities of nasal cilia, diminished spermatic mobility, general reduction of the brain mass, not only from the posterior nostril or visual and hearing systems. Several psychiatric disorders have been associated with Usher syndrome, as schizophrenia, atypical psychosis and frequent depression [1,3]. Other conditions with hearing loss and retinosis pigmentosa are the Waardenburg syndrome, Alport syndrome and Refsum sickness [7]. However, the severity of the clinical picture of these diseases and the age of the onset of symptoms dismiss any relation with our patient.
The visual exploration must include best corrected visual acuity, bio microscopy in slit lamp and ophthalmoscopy. Moreover, complimentary studies include campimetry, macular OCT, ocular fundus pictures, electroretinogram (ERG) [8]. The electroretinogram (ERG) is reported as the ideal study to find retinosis pigmentosa in early stages, and the guide of the Sociedad Nacional de Niños con Sordera recommends that children with sensorineural deafness must undergo this study even if there are no symptoms or signs. Some investigators have proposed, as a routine procedure, to make an ERG in all children with this type of deafness, but it is still under discussion the appropriate age to initiate the screening, which goes from 8 years old and on, or up to before 7 years old in case of congenital deafness. It must be considered that a normal ERG does not exclude the Usher syndrome diagnosis.9,5 Our case presents clear visual affectation in scotopic conditions, and delay in response and intensity to the cones in photopic phase. The prevalence of retinosis pigmentosa in revised patients is 15%, whereas in previous studies without this diagnosis method only reveal a 7 to 10.6%. Changes in the ERG occur even before the clinic manifestation or the signs to the fundoscopy [9]. The genetic study of Usher syndrome allows us to adequately evaluate the use of cochlear implant, genetic council and prenatal diagnosis [10]. Although an earlier study does not represent an extreme change in the treatment or prediction for the affected patients, it can help a genetic council with which the affected family can make decisions, and parents can search for a prenatal diagnosis for future children.
Conclusion
Usher syndrome is a condition that requires a multidisciplinary
attention from the first suspicion. Routine hearing and visual
screening should be made in pediatric patients, since the variability
in the presentation of this syndrome, and the genetic complexity,
important data to conclude its diagnosis at an early age could
escape. A complete study of this disease leads to new adaptation
opportunities of the hearing treatment as well as an adequate
genetic advice for parents and other family members.
For more Articles on : https://biomedres01.blogspot.com/



No comments:
Post a Comment
Note: Only a member of this blog may post a comment.