Pontocerebellar Hypoplasia Caused by De Novo Mutation in PAFAH1B1 (LIS1) Gene
Introduction
The advance of gene sequencing techniques has made it possible to determine the genetic origin of an increasing number of central nervous system malformations which previously did not have a defined etiology. Pontocerebellar hypoplasia (PCH) is a robust example of great variability of phenotypes associated with a specific group of malformations, characterized by atrophic changes of the cerebellar vermis and hemispheres, the ventral portion of the pons and inferior olivary nucleus, often associated with defects in cortical development and derived from mutations in a wide range of genes. To date, at least thirteen subtypes of pontocerebellar hypoplasia with distinct genotypes and phenotypes have been described, but none of them were caused by variants on plateletactivating factor acetylhydrolase IB subunit alpha gene (PAFAH1B1), related to lissencephaly [1]. Lissencephaly is a spectrum of cortical development malformations, characterized by neuronal migration defects, which comprises agyria, pachygyria and subcortical band heterotopia [2,3]. PAFAH1B1, also known as Lissencephaly 1 gene (LIS1), was the first gene identified as being related to lissencephaly, followed by X-linked doublecortin gene (DCX) [4].
Classical lissencephaly (or type 1 - lissencephaly), characterized by the presence of a thick cortex (composed of four abnormal layers) and the absence of other associated brain abnormalities (e.g severe congenital microcephaly, agenesis of the corpus callosum, or cerebellar hypoplasia) [4], is caused by mutations in some specific genes: PAFAH1B1, DCX (in males; in females, DCX mutations are associated with subcortical band heterotopia) and Aristaless-related homeobox, X-linked gene (ARX), in this case, characterized by a three-layered cortex [4]. There are other phenotypes of lissencephaly, associated with microcephaly (called “microlissencephaly”), agenesis of the corpus callosum or even cerebellar hypoplasia. However, until the present moment, there has been no record of mutations in PAFAH1B1 presented with pontocerebellar hypoplasia without cortical malformations, as we describe in this report.
Case Report
The proband is an 8-year-old male, first-child of nonconsanguineous parents, who was born after an uneventful pregnancy, labor and perinatal period (Birth weight: 3,960 g; occipitofrontal circumference: 33 cm). He presented micropenis and surgically corrected bilateral cryptorchidism. He has presented severe global developmental delay, a failure to thrive and deceleration of occipitofrontal circumference growth. Currently he has profound intellectual deficiency, inconstant eye contact, bilateral strabismus, inability to maintain his head up and presents spasticity and dystonia. His occipitofrontal circumference is 47.5 cm (z-score<-3) and fundoscopy is normal. Brain MRI (Figure 1) at 18 months of age disclosed a classic PCH with reduced white matter but normal cortical gyration pattern.
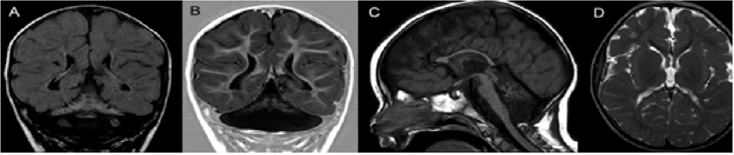
Note: Brain MRI at 18 months of age. Coronal FLAIR (A) and T1 inversion--recovery (B) images disclose a butterfly-type cerebellum, characterized by severe proportional hypoplastic vermis and cerebellar hemispheres. FLAIR image also shows diffuse cerebellar hyperintensity. A sagittal T1-weighted image (C) demonstrates thinning of the corpus callosum, attenuation of the pons, which is almost flat, and a small hypoplastic cerebellar vermis. Axial T2-weighted image (D) shows bilateral reduction of cerebral white matter with unremarkable cortical gyration pattern.
Figure 1: Neuroimaging of Pontocerebellar Hypoplasia.
Genetic Study
Whole exome sequencing was performed in order to identify genetic abnormalities that might be responsible for the clinical and radiological phenotype. No deleterious variants were detected in genes previously associated to PCH, but the patient harbors a missense heterozygous variant p. Arg273Gln (c.818G>A, NM_000430.3; Chr17:2,577,500) in exon 8 of PAFAH1B1, a highlyconserved (PhyloP>2) region and classified by SIFT and Polyphen as deleterious. This variant was neither present in 123,115 individuals from the Genome Aggregation Database (gnomad. broadinstitute.org) nor had been reported before. The variant c.818G>A was confirmed by Sanger sequencing in the index case and his parents were also examined, but it was not present in them.
Discussion
Pontocerebellar hypoplasia (PCH) is inherited as an autosomal recessive or X-linked trait, and it is characterized by profound congenital size reduction of the pons and cerebellum. Several genes have been implicated in PCH, including the autosomal transfer ribonucleic acid (tRNA) splicing endonuclease subunit 2 (TSEN2), TSEN15, TSEN34, TSEN54, O-phosphoserine t-RNA selenocysteine tRNA synthase (SEPSECS), cerebellar atrophy with progressive microcephaly (CLAM), arginyl-tRNA synthetase 2 (RARS2), vaccinia related kinase 1 (VRK1) and the X-linked Calcium/Calmodulin- Dependent Serine Protein Kinase (CASK) [5,6]. Up to now, no dominant inheritance has been associated with PCH. Herein, we report a case of classical PCH, associated with decreased white matter volume, although there is no cortical migration defect. However, molecular testing revealed a novel de novo heterozygous mutation in PAFAH1B1 (LIS1). This gene has been associated with laminar heterotopia and lissencephaly, occasionally combined with PCH [7,8]. In a comprehensive investigation of a large series of PCH, only 60% of cases have their molecular basis unraveled [5]. PAFAH1B1 product plays a critical role in neuronal migration during brain development [7,9].
Haploinsufficiency of PAFAH1B1 leads to neuronal migration defects of variable degrees of severity of the lissencephaly spectrum (OMIM # 607432), including Miller-Dieker syndrome (OMIM#247200). The p. Arg273Gln occurs in the hinge between two of the seven Water Displacement 40th Formula (WD40) domains, which are supposed to form a ring propeller-like structure. No missense mutations in any of the hinge regions of WD40 domains have been reported so far (The Human Gene Mutation Database). The variant Arg273*, leading to premature stop codon, has been reported several times associated with lissencephaly. De novo mutations in coding regions leading to protein structural change occur on average once at every generation [10]. Finding this type of change in a highly conserved 6 gene that is active during central nervous system formation strongly supports its association with PCH. As WES becomes more widespread, the number of genes associated with PCH will probably increase, and PAFAH1B1 might be one of these newcomers. A “new” phenotype for an “old” gene, or an “old” phenotype with a “new” gene?
For more Articles on: https://biomedres01.blogspot.com/



No comments:
Post a Comment
Note: Only a member of this blog may post a comment.