Cations as Molecular Switches for Junctional Adhesion Molecule-A
Introduction
Tight junctions (TJs) are cell-cell promoting structures localized to the apical region of endothelial and epithelial cells. TJs function as barriers, control the paracellular space, and form an apical intramembrane diffusion barrier in the outer leaflet of the plasma membrane, referred to as a fence function [1,2]. TJs are proteic structures represented by a complex mixture of three membrane proteins: claudins (CLDNs), occludin (OCLN), and junctional adhesion molecules (JAMs) [3]. The CLDN family of membrane proteins play central roles in TJ structure and function [3,4]. However, recent literature revealed claudin-independent aspects of the TJ’s function related to JAMs and the TJ’s microenvironment [3]. Additionally, adapter and effector proteins anchor the TJ to the cytoskeleton, indicating its relevance in mechanotransduction [5,6]. Dysfunction of the TJ is relevant to edema, jaundice, diarrhea, inflammatory bowel disease, and metastasis, among other conditions [7]. JAMs comprise a small subfamily of the immunoglobulin superfamily (IgSF) of adhesion receptors. The IgSF members represent a multitude of physiological functions in vertebrate development and homeostasis [8]. The four members of the JAM subfamily (JAM-A, -B, -C and 4) localize to the TJ in a tissue specific manner [9]. Moreover, the first indications for a role of JAM-A in the formation of the epithelial barrier arose from observations in patients suffering from Crohn’s disease (CD) or ulcerative colitis (UC), the two major forms of inflammatory bowel disease (IBD) [10]. Some membrane proteins of the TJ work as receptors pathogenic bacteria and viruses targeting TJ functions [7]; JAM-A in particular engages all reovirus serotypes [11,12].
Studies have shown that loss of JAM-A resulted in a prothrombotic phenotype, and in murine platelets lacking JAM-A there was an impairment of outside signaling [13]. Role of JAMs as signaling molecules as well as cell-adhesion molecules makes them good candidates to coordinate cellular events. Our laboratory has demonstrated that the apical junctional complex (AJC), composed of TJ and AJ substructures, can be modulated by calcium [14]. We demonstrated that just like epithelial cadherin (E-CAD) in the AJ, JAM-A is a calcium-dependent cell adhesion molecule [14]. In our studies we showed that JAM-A, under the influence of calcium, regulates the interplay between the TJ and AJ, thus being a key player in the assembly and function of the AJC [14]. Prota et al. [15], determined that JAM-A forms cis-dimers through crystallography. However, the formation of cis-dimers does not account for cellcell adhesion driven by JAM-A [10,15]. The crystal structure of the extracellular domain of JAM-A attributed the dimer formation to electrostatic and hydrophobic forces, but do not suggest how these can be influenced to alter oligomeric states or adhesive properties [15,16]. Charged and polar groups are responsible for protein properties [17,18]. Modulation of the charges on the amino acids, by pH or by phosphorylation and dephosphorylation or by altering the concentration of cations in the microenvironment, have significant effects such as protein structural changes and switchlike responses leading to protein function [19-21]. An example of how cations affect protein structure and function in cell-adhesion is calcium in CADs [22-24]. Calcium causes the rigidization [25] of the CAD cis-dimers, resulting in further oligomerization of CADs, resulting in cell-cell interactions [26]. Calcium levels in plasma can range from 1.8-2.7 mM [27].
In contrast, intracellular levels of Ca2+ range from 0.3 to 1 mM [28]. In structural studies using E-CAD, the low Ca2+ concentrations (<1 mM) caused the protein to form cis-dimers [24,29]. In the case of high Ca2+ concentration (>1 mM), E-CAD formed trans-dimers [24]. These experiments seem to indicate CADs may form cis-dimers intracellularly but switch to trans-dimers once exposed to the higher levels of Ca2+ in the extracellular microenvironment, leading to cell-cell contacts. Another example of cations’ effect on protein structure was seen in the effect that Zn2+ has on neural cadherin (N-CAD) by mediating cell adhesion in the central nervous system [30]. In synaptic studies it was found that released Zn2+ might have a strong accelerating effect on morphological changes that are involved in long term synaptic plasticity [31]. The synaptic vesicles were found to have a 1 mM concentration of Zn2+ [31]. The presence of JAM-A on the surface of platelets has been described [13,32,33]. The role of cations such as Ca2+ and Zn2+ are well characterized in platelet activation [34-36]. Although our recent report is the only evidence JAM-A is calcium-dependent, the literature recognizes that both JAMs and CADs have similar folding of their extracellular domains [37,38]. Other cations, like magnesium, iron, and copper play physiological roles both in physiology and pathophysiology [39-43].
Addressing this gap in understanding of the role that cations have on JAM-A is better addressed using recombinant proteins and biophysical methods, as reported for E-CAD [44]. In this study we address the following questions: First, do cations affect the oligomerization of JAM-A? To address this question, we purified JAM-A in the presence of different cations using Size-Exclusion Chromatography. Second, do cations have an effect on the conformational change of JAM-A? To answer this question, we used the product from the Size-Exclusion Chromatography to perform Circular Dichroism. Third, do the cations increase the binding affinity of homotypic JAM-A interactions? To answer this question, we performed Surface Plasmon Resonance. Here we present evidence that cations affect secondary, tertiary, and quaternary structure of JAM-A, and ultimately its cell-adhesion properties.
Materials and Methods
Protein Preparation by Cloning, Expression, and Purification of MBP JAM-A
In order to clone, express, and purify the JAM-A protein, we used a synthetic DNA gBlock obtained from IDT (Integrated DNA Technologies, Iowa City, IA, US). The gBlock was amplified with forward and reverse primers using PCR (Figures S1 & S2). Maltose binding protein (MBP) was cloned N-terminal of JAM-A using NcoI and NdeI sites on plasmid pET28a (Millipore Sigma, Burlington, MA, US). An amino acid sequence of these plasmids is provided in Figure S3. Plasmids were transformed in SHuffle bacterial cells [45,46]. We followed our previously described protocol for protein expression of MBP JAM-A [47]. Lysis buffer (0.5 M EDTA, 500 mM NaCl, 30 mM Tris pH 7.4) followed by French Press. Samples were loaded onto the Thermo Spectronic French Pressure Cell Press Model FA-078. Lysis was performed at 1500-2000 psi and the lysate was collected in a new 50 mL tube. The lysate was centrifuged for 30 minutes at 11,000 g using a F15-8x50cy rotor from Thermo Scientific. The supernatant was decanted into a 50 mL Eppendorf tube containing Amylose resin (cat# E8021L) from New England Bio Labs and incubated while rotating for 1 hour at 4°C. Column chromatography was performed to collect the protein from the amylose beads. Then the column was washed with 100 column volumes of wash buffer containing 500 mM NaCl and 30 mM Tris pH 7.4. The elution was incubated for 3 minutes each time. The eluate was concentrated by using the Microsep Advance with 10k Omega centrifugal devices (Reference # MCP010C41) from Pall Corporation and centrifuged at 10,000 RPM for 10 minutes until reaching 2 mL of final elution volume.
Size-Exclusion Chromatography (SEC)
Determination of the shift of oligomerization was performed using 0.250 mg of protein in one of the solutions of 30 mM HEPES, 100 mM NaCl with 2 mM of each cation (calcium, copper, magnesium, zinc, phosphate, iron II, or iron III) as chloride salt. The protein was incubated in the buffer with the cation of interest for 2 hours. The sample was injected into the injection valve of the NGC Chromatography System (Bio-Rad) with a syringe. The elution peaks and change in the area under the curve was calculated with SEC ChromLab 4.0 software (Bio-Rad).
Circular Dichroism Spectrometry
Circular dichroism (CD) measurements were performed on Spectropolarimeter Model 420 (AVIV Biomedical Inc., Lakewood, NJ USA). Changes in ellipticity were performed from 250 nm to 190 nm using 20 second scans at a concentration of 100 μM protein in a 10 mm QS glass cuvette at 22°C. The secondary structure which consisted of alpha helix, antiparallel, parallel, turn, or other was determined by using the online Bestel circular dichroism analysis software: https://bestsel.elte.hu/index.php.
SDS-PAGE Assay
Protein samples were prepared as follows: 1 μg of either heat treated or untreated MBP, JAM-A with the various cations were electrophoresed on 8% SDS-PAGE gel with loading dye containing 1% SDS, 0.125 M Tris (pH 6.8), and 40% glycerol. Protein bands were visualized after staining with Coomassie Brilliant Blue for 2 hours. The Coomassie Brilliant Blue was distained with distain buffer (40% MeOH, 10% Acetic Acid, 50% H2O) for 2 hours.
Native Gel Assay
Native gel consisted of 15% resolving gel recipe containing 2.5 mL ultra-pure H2O, 5 mL 30% polyacrylamide/Bis Solution (29:1), 2.5 mL 1.5 M Tris HCl pH 8.8, 5 μL TEMED and 50 μL 10% APS (Ammonium Persulfate) added as the last step. The 5% stacking gel consisted of the following: 2.3 mL ultra-pure H2O, 62 μL 30% Acrylamide/Bis Solution (29:1), 1 mL 0.5 M Tris HCl pH 6.8, 5 μL TEMED, and 30 μL 10% APS (added last to allow for gel solidification to occur). Protein was loaded 1 μg per well and ran at 100 volts for 110 minutes. Protein bands were visualized after staining with Coomassie Brilliant Blue for 2 hours. The Coomassie Brilliant Blue was distained with distain buffer (40% MeOH, 10% Acetic Acid, 50% H2O) for 2 hours.
Surface Plasmon Resonance (SPR)
We employed Open SPR by Nicoya Lifesciences to assay protein-protein associations of JAM-A with the incubated cations. SPR is an optical effect that can be utilized to measure the binding of molecules in real time without the use of labels. SPR instruments are primarily used to measure the binding kinetics and affinity of molecular interactions. SPR can be used to measure interactions such as the binding between two proteins, a protein and an antibody, DNA and a protein, and many other molecules. SPR can be thought of as the following equation: Analyte + Ligand ßà Analyte-Ligand (Complex). This equilibrium equation shows that not all of the ligands will be bound to the protein. When the ligand is bound to the protein forming the complex this is considered to be Kon (M-1s-1) or the speed of association. Koff is the speed of dissociation (s-1). A final calculation enables the experiment to reveal the Analyte-Ligand affinity: KD=Koff/Kon=([Analyte]x [Ligand])/[Analyte-Ligand] Complex. The KD is the dissociation constant where half the ligand binding sites of the protein are bound to the ligand and half of the ligand is not bound to the protein at equilibrium. Thus, a smaller KD value means that the analyte and the ligand have higher binding affinity for one another (Nicoya Lifesciences, user’s manual). In our experiments, the ligand and the analyte is MBP JAM-A. To determine the binding affinity, we used 0.050 mg of each protein as a ligand into the Sensor Carboxy Chip, for coupling to any amine group on the ligand (Nicoya Lifesciences).
The proteins were immobilized in the Carboxy Sensor Chip through the exposed primary amine groups that are found in the lysine residues and at the N-terminus. These primary amines form covalent bonds with the carboxyl surface after it is activated by the EDC/NHS (Nicoya Life Sciences) [48]. The blocking step (manufacturer’s buffer) followed by 200 µL of 1 M sodium caprate was administered to disrupt the preformed protein-protein interactions [49]. Triplicate injections of the analyte protein were made in the following concentrations: 12.5 µg, 25 µg, 50 µg and 100 µg per 200 µL injection. Caprate injections were performed after each analyte interaction to be sure that there were no other interactions occurring before the next analyte injection was performed. After conducting the experiments, the close curve fitting to the sensograms was calculated using global fitting curves using the 1:1 Langmuir binding model. The data was retrieved and analyzed with Trace Drawer software (Kitchener, ON, Canada). SPR for each sample was performed in triplicate and analyzed using the Trace Drawer software (Kitcherner, ON, Canada) according to the recommendations by Nicoya (Kitchener, ON, Canada).
Tissue Culture
HEK 293 cells (ATCC catalog# CRL-1573) were cultured following standard procedures in DMEM media with 10% FBS. CAL 27 cells (ATCC catalog# CRL-2095) were cultured using DMEM-F12 media with 10% FBS. We followed ATCC culture guides (https://www.atcc.org/resources/culture-guides).
Confocal Microscopy
A confocal microscope (Olympus Fluo View FV1000) was used to observe the transfected HEK 293 cells. The microscope is equipped with an argon laser with excitation light at wavelengths of 405nm, 458 nm, 488 nm, and 515 nm. In addition, it provides green Helium-Neon and red Helium-Neon laser sources with respective excitation wavelengths of 543 nm and 633 nm. The cells were observed and imaged at 20x magnification using standard procedure. The Olympus Fluo View FV1000 software was used to obtain images of the samples.
ATP Proliferation Assay
Using Cal 27 cells we performed proliferation assays using ATPlite Luminescence Assay System (PerkinElmer, Akron, Ohio) following the manufacturer’s instructions.
DNA and siRNA Transfections
HEK 293 cells were transfected with plasmids following standard procedures [49] in 6-well plates with 2 ug of DNA (Figure S3) per well using jetPRIME transfection reagent (Polyplus, New York), according to manufacturer’s instructions. CAL 27 cells (2×105) were seeded in 12-well plates and transfected after 24 h with 30 nM siRNA duplexes using Jet Prime transfection reagent, according to the manufacturer’s instructions. siRNA duplex oligonucleotides were synthesized by Integrated DNA Technologies (IDT, Coralville, Iowa), control siRNA (IDT cat#51-01-14-03); against human E-CAD [50], against human JAM-A [51].
Statistical Analysis
Student’s t-tests were applied for comparisons. Data are expressed as mean±SD. The significance threshold was 5% (*P<0.05). All experiments were repeated four times.
Results
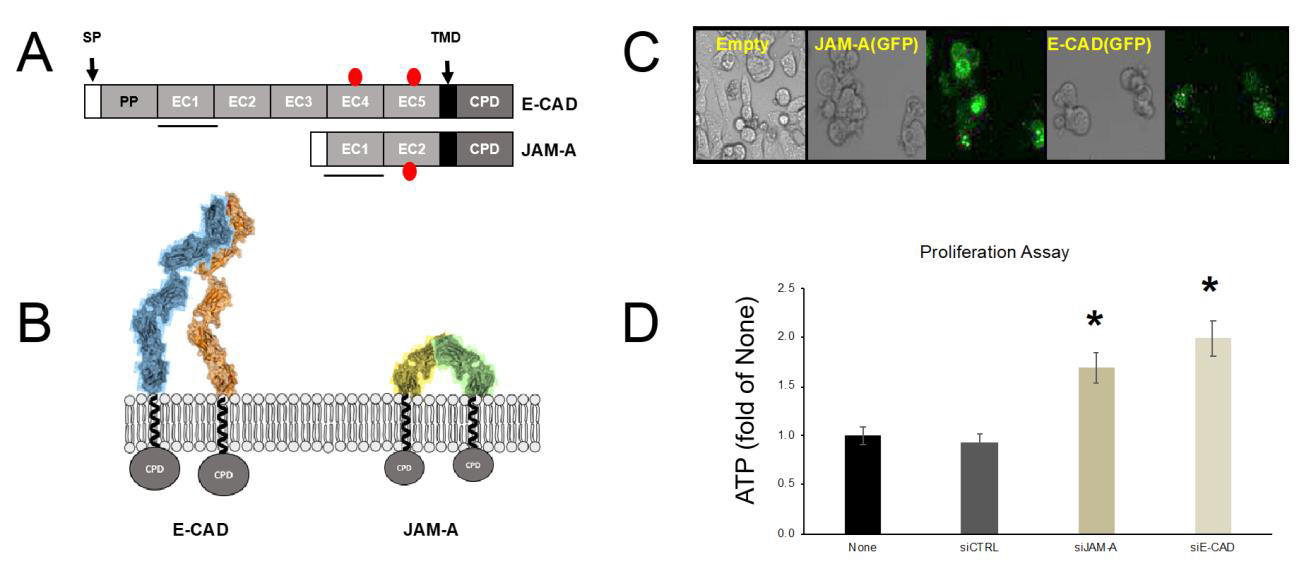
Figure 1: Similarities between JAM-A and E-CAD in a calcium-free environment.
A. Schematic structure of E-CAD and JAM-A. SP, signal peptide; PP, pro peptide; EC, extracellular domains of Ig folding; TMD, transmembrane domain; CPD, cytoplasmic domain. Red circles denote glycosylation sites, the solid black line indicates the binding Ig domain for cis- and trans interactions.
B. Models of cis-dimer formation based on crystallographic studies. Two E-CAD monomers interact through EC1 (blue and orange). JAM-A in the absence of calcium forms a U-shaped dimer (green and yellow monomers).
C. HEK 293 cells transfected with Empty vector (pcDNA 3.1), pcDNA 3.1 JAM-A(GFP), or pcDNA3.1 E-CAD(GFP).
D. ATP proliferation assay of CAL 27 cells. Gene silencing was achieved by siRNA primer transfection. Untreated cells (None) were not transfected; siCTRL, cells were transfected with non-specific primers; cells were transfected with specific primers to knockdown JAM-A (siJAM-A) or E-CAD (siE-CAD). The (*) denotes statistically significant (p<0.05) compared to siCTRL.
Proteins are organized in families. The categories depend on primary amino acid sequence homology, conserved residues, domain folding, and other tertiary and quaternary structural elements. The biggest protein family is IgSF with close to 750 members [52,53]. Among the functions observed for the different subfamilies of the IgSF we find pattern recognition and cell-adhesion molecules [54,55]. CADs are calcium-dependent cell-adhesion molecules that form the membranal structure of the AJ. Calcium-free CAD’s form cis-dimers and do not result in cell-cell adhesion. JAM-A is a protein from the IgSF that also has cell-adhesion properties. JAM-A’s crystal structure [15,56], was obtained in the absence of cations. This calcium-free structure results in a U-shape cis dimer. We proceeded to design a few simple experiments to characterize structure and function similarities between E-CAD and JAM-A. Figure 1 indicates important similarities between E-CAD and JAM-A in a calcium-free environment. E-CAD and JAM-A form cis-dimers in the absence of calcium. Both E-CAD and JAM-A use EC1 for binding and glycosylation occurs in domains away from the binding site. Figure 1C reveals that upon transfection of E-CAD or JAM-A, as GFP fusion proteins, resulted in a rounding phenotype in HEK 293 cells, which is a feature described in the literature for overexpressing cells that affect morphology [57,58].
A final level of similarity is revealed by the ATP proliferation assay Figure 1D. CAL 27 cells (epithelial tongue Squamous Cell Carcinoma) contain a very simple TJ, composed of JAM-A, CLDN1, and OCLN. Additionally, as an epithelial cell-derived product CAL 27 cells express E-CAD. In the literature, the knockdown of E-CAD in human ovarian cancer cells is related to increased proliferation [50]. In our experiments with CAL 27 and siRNA specific for E-CAD or JAM-A, we observed that both knockdown experiments resulted in increased cell proliferation when compared to control (non-specific) siRNA primers. Considering the above-described similarities between E-CAD and JAM-A we hypothesized that JAM-A could be calcium-dependent. An interplay between the TJ and AJ has been described in a recent review [59]. Our laboratory demonstrated recently that Ca2+ acts as a molecular switch affecting primarily JAM-A to coordinate the participation of both TJ and AJ in the assembly and function of the AJC [14]. Cations act as switches in the CAD subfamily [60,61]. Most CADs are recognized as calciumdependent [22,62], and others are recognized to be magnesiumdependent [63], nickel-dependent [64], and zinc-dependent [31]. Finally, CAD desmosomes adhesive properties are decreased by calcium [65]. At least 30% of proteins bind metal ions. Bound ions are essential for protein folding, subunit assembly, interaction with other macromolecules, and protein function [66].
In this study we determined that cations influenced the secondary structure, binding affinity and oligomerization of MBP JAM-A. Our previous work [47] showed that each of the four JAM proteins that form TJs had a unique tertiary and quaternary structure, but they had similar secondary structures. We also determined that JAMs are involved in homotypic or heterotypic interactions [47]. Our previous studies were performed in the presence of Phosphate saline solution (PBS) [47]. We did not address whether changes in ionic interactions due to buffer conditions, including cations, could affect JAM protein properties. Similarly, the study by Prota and colleagues [15] reported the crystal structure of the extracellular domain of JAM-A but did not address whether cations would affect dimerization and higher order oligomerization as established for CADs [44]. In this study, we designed experiments to determine the effect of different cations on MBP JAM-A. We mainly observed different oligomeric forms depending on the cation to which it is exposed in solution. Changes in aggregation have been demonstrated for proteins and nanoparticles when exposed to different cations or different ionic strengths [16,67]. We determined that the different cations changed the secondary, tertiary, and quaternary structure of MBP JAM-A.
Prediction of Cation Binding Sites in JAM-A
In order to predict the cation-binding sites the crystal structure of the protein of interest must be known [68]. Therefore, to perform the prediction we used JAM-A crystal structure PDB ID 1NBQ and the MIB: Metal Ion-Binding Site Prediction and Docking server (http://bioinfo.cmu.edu.tw/MIB/). The metal cations used in this docking server were: Ca2+, Mg2+, Cu2+, Zn2+, Fe2+, and Fe3+. The software gave us ranking scores of potential cation binding sites for JAM-A, where the cation docking sites with the highest scores represent the probability of being a cation binding site [68,69]. The docking sites with the highest scores were used for visualization of the different cations using UCSF Chimera (Figures 2A-2F). The lesser ranked predicted sites can be found in the supplementary file, (Figure S4). The amino acid sequence of JAM-A was used to label the potential docking sites with the highest score for the cations (Figure 2G). The MIB server did not identify sites in MBP (PDB ID 1NL5), data not shown. All the binding sites correspond to unstructured amino acids in the structure of JAM-A [15]. To validate the utility of the data obtained from the MIB webserver, we predicted calcium-binding sites of E-CAD (Figure S5). The MIB server adequately predicted the sites observed in E-CAD calcium crystal structure [23]. This result highlights the relevance of MIB as a predictive tool for JAM-A. After the determination of cation docking sites, we purified the MBP JAM-A protein to perform the experiments in the next section.
Expression System, Cloning, And Purification in E. Coli
In this study we recombinantly expressed the two extracellular immunoglobulin domains of JAM-A. We used Maltose Binding Protein (MBP) as a fusion partner to allow for the generation of high yield of proteins, and to maintain consistency with our previous work [14,47,70-72]. The pET28-MBP-JAM-A (Figure S3) plasmid was expressed in the SHuffle T7 bacterial strain to allow for the proper folding, disulfide formation and cytoplasmic expression of the JAM-A extracellular protein [45]. Following Amylose resin purification, Size-Exclusion Chromatography was carried out with 2 mM of each cation (Cu2+, Ca2+, Mg2+, Zn2+, Fe3+, Fe2+) in HEPES buffer (100 mM NaCl, 30 mM HEPES at pH 7.4). The protein had high yields with a purity of >95% (Figure 3B). Additionally, oligomerization changes of JAM-A were observed for the different cations, which was shown using a native gel (Figure 3C) without SDS, which maintained the protein in its native structure and allowed oligomers to be observed.
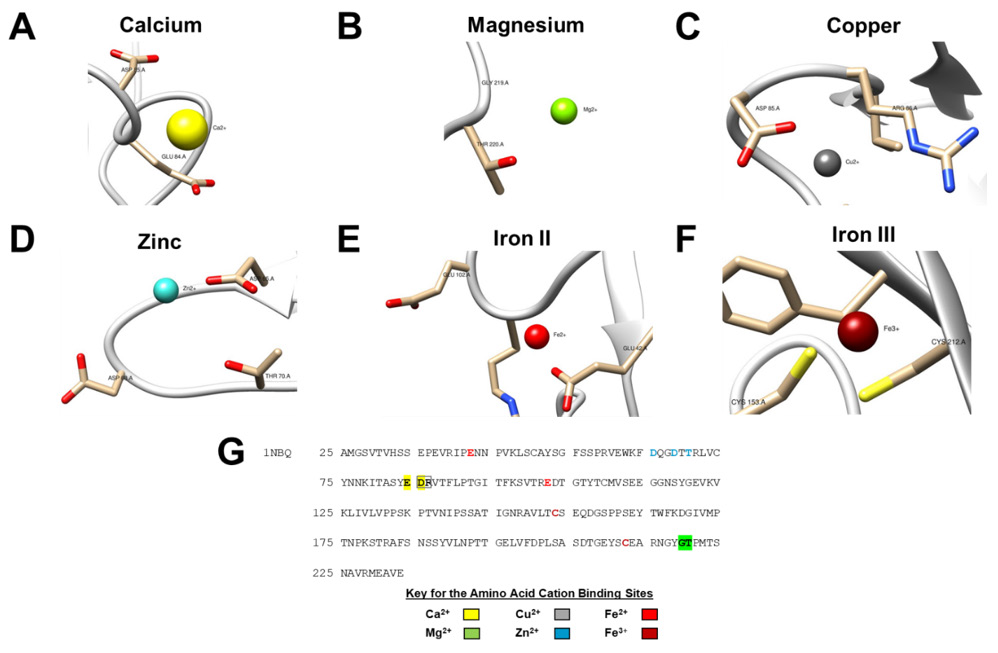
Figure 2: Prediction of cation-binding sites in JAM-A (PDB ID 1NBQ).
A. Ca2+ binding sites were predicted to be ASP85 and GLU84.
B. Mg2+ predicted binding sites were GLY219 and THR220.
C. Cu2+ predicted binding sites were ASP85 and ARG86.
D. Zn2+ predicted binding sites were ASP65, ASP68 and THR70.
E. Fe2+ predicted binding sites were GLU42 and GLU102.
F. Fe3+ predicted binding sites were CYS153 and CYS212. Predicted binding sites were generated by the MIB webserver (http://bioinfo.cmu.edu.tw/MIB/). Models were visualized using UCSF chimera [70,71]
G. Amino acid sequence of the JAM-A crystal structure PDB ID 1NBQ shows the cation binding sites labeled according to the cation color in panels A-F.
Determination of Protein Shift by Size Exclusion Chromatography
The function of cell-adhesion molecules such as JAM-A depends on their ability to dimerize or multimerize. Size-exclusion chromatography (SEC) has been established as a powerful tool to determine oligomeric state and changes in oligomeric state [73] and has been used to study protein denaturation [74]. Further purification of MBP JAM-A was performed using SEC. Size-exclusion peak shift and oligomerization were determined using 2 mM of each cation chloride salt (Cu2+, Ca2+, Mg2+, Zn2+, Fe3+, Fe2+) in HEPES buffer (100 mM NaCl, 30 mM HEPES at pH 7.4). Among all the cations, we determined that the greatest shift from dimer to oligomer was observed in the presence of Zn2+, which was not observed with the rest of the cations (Figure 3). SEC columns cannot accurately predict size beyond MBP JAM-A tetrameric form. The term oligomer was used for Zn+2 and it could represent an octamer or higher order of quaternary organization. Our data suggests a correlation between the formation of oligomers and the increased homotypic binding of MBP JAM-A. Furthermore, we investigated whether the increase in binding was the result of changes in the secondary structure of MBP JAM-A. To determine whether cations affected JAM-A’s secondary structure as evidence of changes in self-association, we used Circular Dichroism.
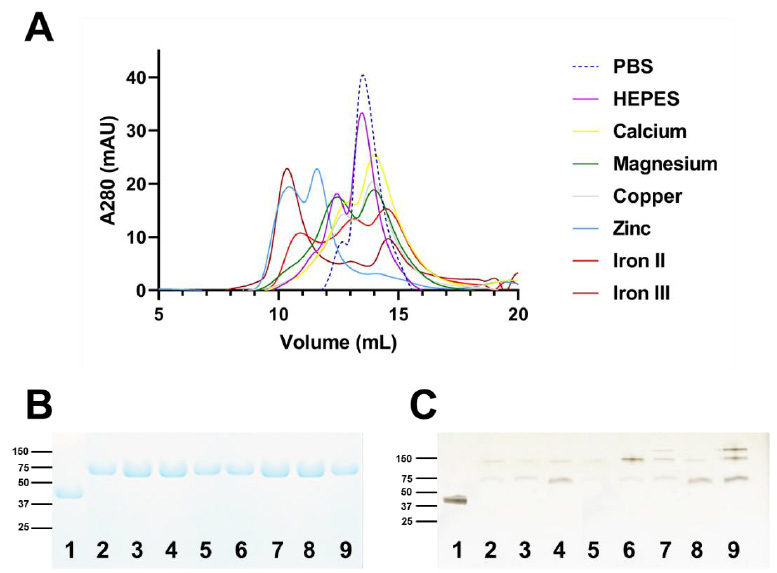
Figure 3: The effect of examined cations on MBP JAM-A oligomerization.
A) The shift of MBP JAM-A with the cations was seen as follows:
1) Zn+2 produced multimers,
2) Ca+2 produced dimers and slight monomers.
3) Mg+2 produced dimers and slight monomers,
4) Phosphate buffer produced dimers,
5) Cu+2 produced dimers and shifted to a slight tetramer.
6) HEPES buffer produced a dimer and slight tetramer.
B) SDS PAGE of purified MBP JAM-A exposed to different cations. All of the proteins in the presence of 2 mM cations, where purified by size-exclusion chromatography were determined to be JAM-A at > 95% purity. Maltose Binding Protein (MBP) was used as a control in lane 1. All other samples were MBP JAM-A with different cations following size exclusion, lanes 2 to 9, in the following order: PBS, HEPES, Ca+2, Mg+2, Cu+2, Zn+2, Fe+2, Fe+3.
C) Native gel of purified MBP JAM-A exposed to different cations. MBP JAM-A homotypic interactions were observed when exposed to the different cations. Samples were loaded in the same order as in Panel A, with MBP as control. For panels B and C, the relevant molecular weights are shown in the ladder.
Cations Effect on JAM-A Secondary Structure by Circular Dichroism (CD)
In Figure 4 we present the CD of both MBP (unfused) and MBP JAM-A. Additionally, Figure 4 contains two tables corresponding to the structural composition of the proteins analyzed; α-helix, β-sheet content, as well as Other, a combination of turns and unstructured loops. The initial analysis demonstrate that buffer composition (PBS or HEPES) can modify the structural distribution of proteins as reported in other instances [75,76]. Our results reveal that changes in secondary structure (Figures 4A & 4B) are not sufficient to induce changes in tertiary structure of MBP since in SEC we did not observe changes to its elution volumes (data not shown). When compared the changes to secondary structure of MBP JAM-A (Figures 4C & 4D) and its tertiary structure (Figure 3C) we observed two different trends. To illustrate our observations, we will cite work in which it is observed that calcium induces rigidization of E-CAD, leading to trans-oligomerization corresponding to the structural form that results in cell-cell interactions [24,77,78]. Rigidization or a decrease in unstructured regions is observed with calcium, magnesium, and zinc (Figure 4D), where initial other content was 35.3% with HEPES alone and less than 5% with the mentioned cations. Zinc seems to drive oligomerization to the greatest extent from this group (Figures 3A & 4D) while favoring an increase in both α-helix (18.9%) and β-sheet (15.0 %) content. In the case of Iron II and Iron III, both increased MBP JAM-A oligomerization (Figure 3A) but did not modify the unstructured content of the protein (Figure 4D). Copper seems to be an intermediate case (Figures 3A & 4D).
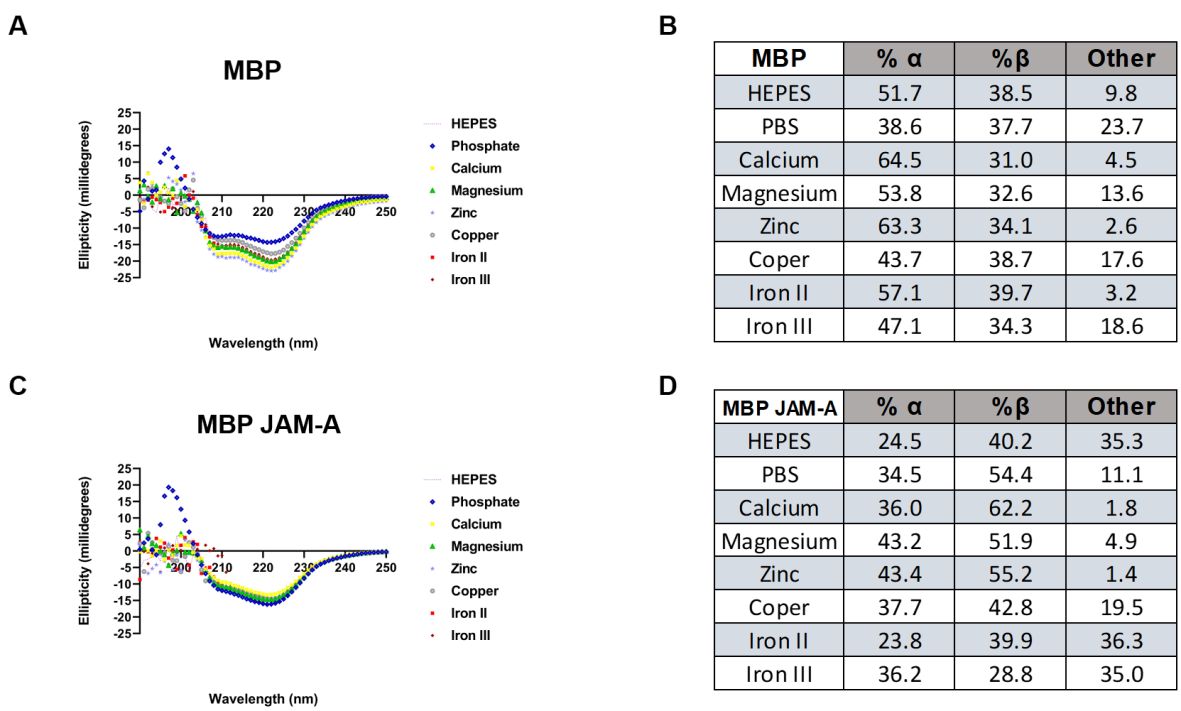
Figure 4: Circular Dichroism of MBP and MBP JAM-A exposed to different cations.
A) Changes to the secondary structure of MBP exposed to different cations at 22 °C.
B) The changes in the secondary structure from Panel A, are shown in the table highlighting percentages of Alpha, Beta, and Other (turns and unstructured loops).
C) Changes to the secondary structure of MBP JAM-A when exposed to different cations at 22 °C.
D) The changes of the secondary structure, seen in Panel C, are shown in the table highlighting percentages of Alpha, Beta, and Other (turns and unstructured loops). Data presented in the tables was analyzed using the BeStSel server (https://bestsel.elte.hu/index.php).
Our results suggest that cation exposure of MBP JAM-A results in secondary structure changes and may be related to tertiary structure changes. These changes produce oligomeric states beyond the formation of dimers observed both with HEPES and PBS buffers (Figures 3 & 4). Furthermore, comparing calcium effects on E-CAD, Koch and colleagues studied changes to CD spectra of E-CAD extracellular domains 1 and 2 [23]. The authors suggested that calcium binding to E-CAD resulted in conformational changes that affected the rigidization of the protein structure in preparation for binding [23]. Finally, the structural landscape of E-CAD [79] includes the formation of cis-dimers in multiple conformations (x-dimer and S-dimer) that can be transitioned by calcium acting as molecular switch to the trans-oligomeric state, triggering cell-cell interactions through rigidization [44,79]. The NMR structure of E-CAD [78] also suggests that the structure is largely dynamic, depending on protein concertation and calcium binding. This can be equated to protein expression and abundance, and the electrostatic contributions of the environment. Other examples of the effects of cations have on the changes in protein structure and stability are seen in calcium-binding protein 1, prothymosin α, avian thymic hormone, hepatitis C virus NS3 protease, and calcium and integrin- binding protein [80-85]. Avian thymic hormone (ATH), which is expressed in the chicken, consists of two β-parvalbumin isoforms [81, 83]. It has been found to have cation-binding sites with the following dissociation constants: 4-10 nM for Ca2+ and 40-80 µM for Mg2+ [82].
Studies to determine the effect on the secondary structure of ATH showed that Ca2+ did not produce distinguishable changes in secondary structure, but Mg2+ exposed the hydrophobic regions to the solvent, shown by fluorescence emission spectra [84,85]. In the case of calcium-binding protein 1 (CaBP1), it has been found that it not only binds to three Ca2+ ions, but also binds to one Mg2+ ion with a dissociation constant of 300 µM [86]. This is linked to the conformational changes of the CaBP1 protein, because when exposed to cations it formed a dimeric conformation, but in the absence of cations it formed a molten globule-like structure, shown by dynamic light scattering [86]. This suggests that cation binding induces homodimerization and structural stability of CaBP1 [86]. This leads to CaBP1 promoting the opening of the L-type Ca2+ channel when compared to Calmodulin (CaM) [87-89]. Calcium- and integrin-binding protein (CIB) is another example of a protein with Ca2+ -binding sites. Studies have shown that CIB has an affinity for Ca2+ with dissociation constants of 0.5 and 2 µM [90]. Therefore, Ca2+ leads to a conformational change that stabilizes the secondary and tertiary structures of the protein. Through CD experimentation it was found that CIB had a helical content and random coil structures. This was caused by the exposure to Ca2+, which affected the α-helical content, unstructured protein regions and tertiary structure of the protein [90]. To address the effect that cations had in the homotypic binding (self-binding) of MBP JAM-A, we performed Surface Plasmon Resonance (SPR).
Determination of the Effects of Cation on MBP JAM-A Homotypic Interactions By SPR
The homotypic interaction of MBP JAM-A was observed to increase with exposure to cations. The largest effect was due to Zn2+ when compared to HEPES, and PBS alone. Based on these results (Figure 5), KDs for homotypic interactions were ranked from greatest to least binding: Zn2+ > Cu2+ > Mg2+ > Ca2+ > Fe3+ > PBS > Fe2+ >HEPES. Our results mirrored the shifts found with SEC (Figure 3A) and suggest that affinity binding between these proteins changed depending on the cation it was exposed to. The protein experienced conformational changes, as determined by CD (Figure 4), which resulted in changes in binding, measured by SPR (Figure 5). The increased binding affinity for homotypic MBP JAM-A could be based on the conformational change that the cations produced in the secondary structure of the protein. Zinc produced the highest binding affinity, compared to the other cations and HEPES, which may originate on the conformation of the β-sheet, α-helix content (Figure 4). MBP JAM-A in HEPES had a constant of affinity of approximately 48 µM, but when exposed to Zn2+, the affinity increased to 28 nM. Higher affinity is related to an increased in buried contact surface [91], which is in agreement with Zn2+ ‘s effects on MBP JAM-A, increased oligomeric state and increased homotypic affinity. The metal cation binding sites of the protein may have become closer when bound, which could lead to conformational changes [92-94] in the MBP JAM-A protein. Calcium and magnesium had different binding affinities that caused different conformational changes in the binding of target proteins. The different cation binding sites predicted from JAM-A (Figure S4) may result in a regulatory switch leading to the conformational changes and ultimately contribute to the TJ formation and cell-cell adhesion [95,96].
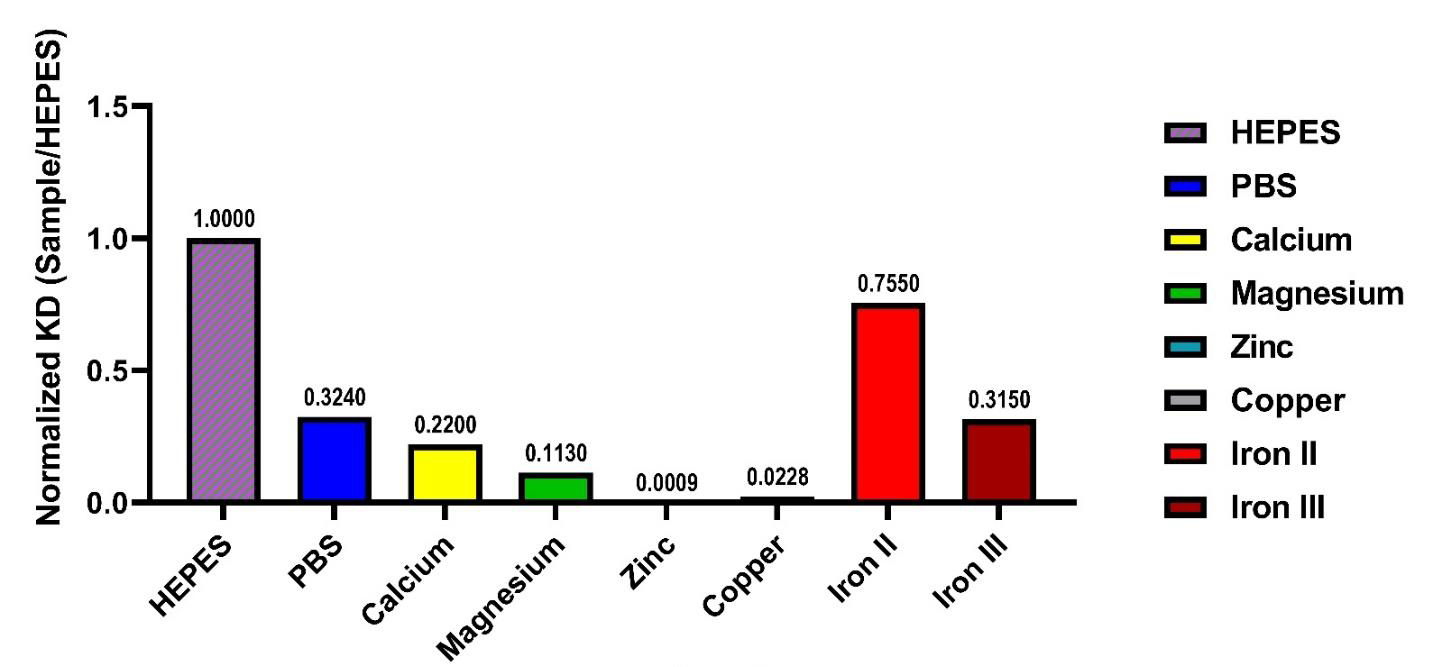
Figure 5: Cations affect homotypic binding of MBP JAM-A. Binding affinity (KD) of MBP JAM-A under the effect of different cations was normalized to the value obtained with HEPES. The ranking of affinities is presented in the following order, with the smallest value in the graph as the highest binding affinity and the largest number as the lowest binding affinity: Zn2+ > Cu2+> Mg2+ > Ca2+ > Fe3+ > PBS > Fe2+ >HEPES.
We present the data obtained from studying JAM-A under the influence of different cations. See Table 1, containing Ka (association constant), Kd (dissociation constant), and KD (binding affinity calculated by Ka/ Kd). The homotypic binding of JAM-A was affected by cations. When exposed to Zn2+, the constant of affinity was the highest, as shown by SPR (Table 1). Overall, the changes in conformation of JAM-A, driven by the cations, would orchestrate the binding and oligomeric state of JAM-A. Taken together, this suggests that JAM-A may play a role in how TJs are assembled, leading to changes in binding and oligomerization that can either increase or decrease the TJ function. Our recent report is evidence that calcium-dependent JAM-A is a factor in interplay between the TJ and AJ for assembling AJCs [14]. This novel finding suggests that the TJ components are tightly regulated by changes in extracellular cation concentrations. Another consequence could be that the ultra-structure of the TJ may be controlled by cation concentration or other microenvironment events that alter the electrostatic properties surrounding it. Our previously cited study also shows that other components of the TJ, namely CLDN1 and OCLN, are not influenced by calcium [14]. Studying cations effect on other JAMs is also key to understanding the interconnection between TJ and AJ. Finally, considering that the crystal structure of JAM-A has been determined, future crystallographic studies may include cations of interest.
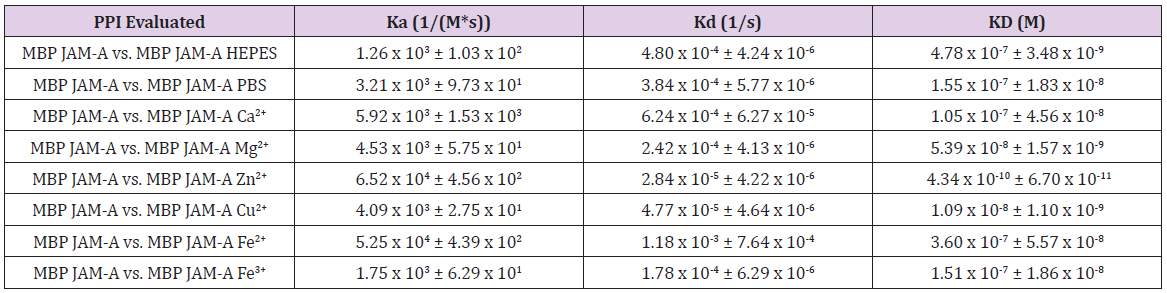
Table 1: Surface Plasmon Resonance (SPR) analysis of MBP JAM-A when exposed to different cations. All experiments had a Chi2 value less than 10% of Rma [95,96]. The values obtained by SPR are Ka (association constant, that measures the rate at which the two partners bind during the association phase), Kd (dissociation constant that measures the rate at which the protein called the anylate separates from the ligand attached to the sensor chip), and KD (binding affinity calculated by Ka/ Kd).
Effect of Cations on JAM-A Associated Morphology and Proliferation
Overexpressing JAM-A in HEK 293 cells results in cell rounding (Figure 1). Our JAM-A construct was tagged with C-terminal GFP (Figure S3). We observed that 48 hours post-transfection and in the absence of cations, cells remain mostly round and do not aggregate. This result has been shown in studies where the knockdown of JAM-A accelerates the proliferation and migration of human keratinocytes [51]. Our data indicates that different cations increase cellular aggregation (Figure 6, panels 2-4). For example, the influence of zinc on JAM-A(GFP)-transfected cells aggregated the most. In Fig. 6B we show the cellular aggregation between two adjacent cells expressing JAM-A (GFP). In the literature, E-CAD overexpression results in similar cell-cell adhesion structures [77]. We obtained similar results using our E-CAD(GFP) construct in HEK 293 cells exposed to calcium (Figure S6).
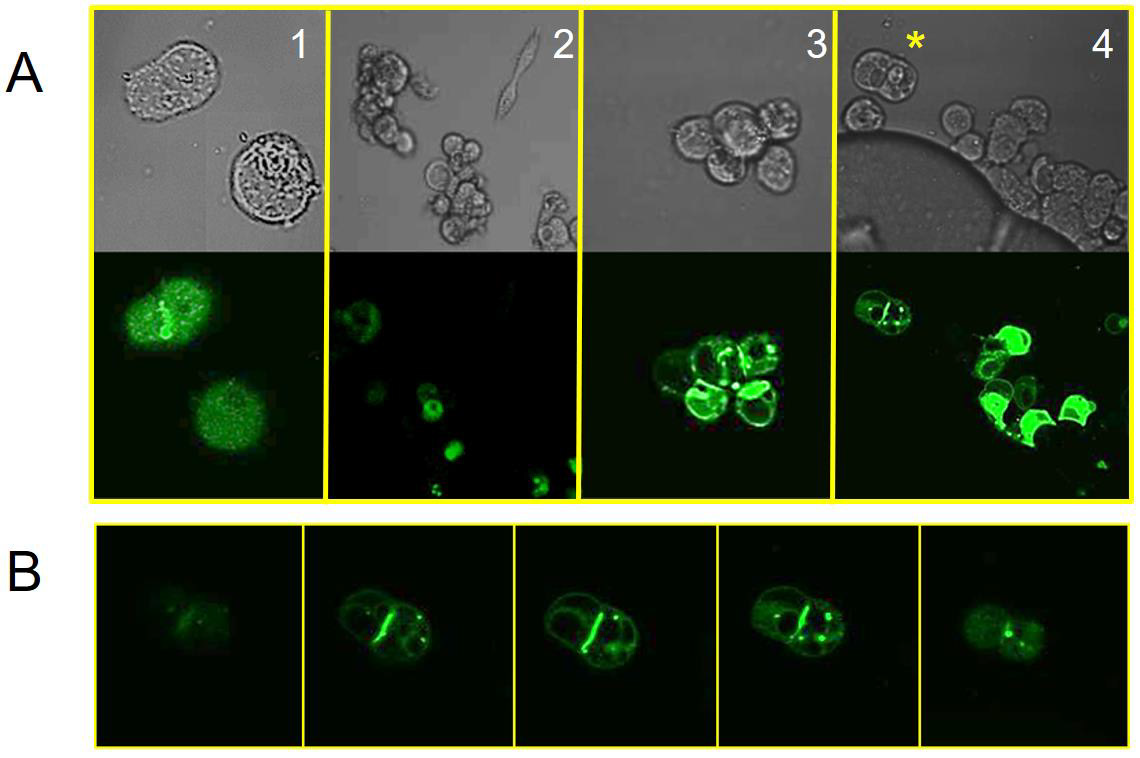
Figure 6: Morphology changes to HEK293 cells transfected with JAM-A(GFP).
A. HEK 293 cells were transfected with JAM-A(GFP) plasmid. 24 hours post-transfections cells were exposed to buffer (panel 1) or cations (calcium, panel 2; magnesium, panel 3; zinc, panel 4). Below each bright field image is the corresponding capture using GFP/FITC filter. The (*) denotes the 2 cells used in the next panel.
B. The 2-cell structure from panel 4 (zinc) was further studied using Z-stack (4 nm increments). This gallery shows 5 optical sections taken from the whole length of the cell, top to bottom (left to right). Comparing the bright field image with this sequence of images we observe that JAM-A(GFP) is distributed through the interface.
Conclusion
JAM-A is a protein important to the formation of TJs but very little is known about the role that cations play in regulating its structure, oligomeric state, and binding properties. In this study, we determined that cations affect the secondary, tertiary, and quaternary structure of JAM-A, and its binding affinities in homotypic interactions. Our contribution brings to light the key role that cations play in regulating the homotypic interactions of JAM-A [15,47,97]. The exposure of JAM-A to different cations resulted in changes in the secondary structure, which may be linked to the effects on binding as seen in our SPR analysis. Based on our results, we present a graphical ranking (Figure 7) that highlights the effects that cations have on the homotypic binding affinity of JAM-A. These cations, of physiological importance can reach high concentrations in the plasma or extracellular fluids and thus act as molecular switches regulating JAM-A homotypic interactions and ultimately cell adhesion. The role that cations may play in the secondary structure, oligomerization, and binding of JAM-A could influence TJ formation in different cell types and tissues and orchestrate the interactions between TJ and AJ. Authorship Contributions: Christopher Mendoza: protein expression and purification, Coomassie, Circular Dichroism, Surface Plasmon Resonance, experimental design, experimental performance, data interpretation and protein modeling. Sai Harsha Nagidi: Surface Plasmon Resonance, data interpretation. Keegan Peterson: Confocal Microscopy and imaging analysis. Dario Mizrachi: Protein design, protein expression and purification, experimental design, data collection, and data interpretation.
Figure 5: Graphical ranking of cations affecting the homotypic binding affinity (KD) of JAM-A. In vitro (recombinant protein) ranking suggests that cations affect JAM-A binding, with the greatest amount of binding at the top and decreasing toward the bottom. As JAM-A is part of the TJ, we suggest that its contribution to the strength of the barrier function will be modified by the presence of cations in the environment.



No comments:
Post a Comment
Note: Only a member of this blog may post a comment.