Prenataly Diagnosed Bilateral Congenital Cystic Adenoid Malformation, Class 2, a Case Report
Introduction
The term congenital lung malformation cover a wide range of disorders: congenital cystic adenomatoid malformation (CCAM), intra- and extra-lobar pulmonary sequestration (PS), bronchogenic cysts, congenital lobar emphysema and bronchial atresia. Theese malformations account for 5% of all congenital abnormalities, have a cumulative incidence of 30 cases per 100,000 individuals, and thus are considered rare disorders [1]. Congenital cystic adenomatoid malformations (CCAM) are developmental abnormalities of the lung. It was reported as a separate entity in 1949 by Chin and Tang. They are generally characterized as benign hamartomatous or dysplastic overgrowth of terminal bronchioles with a reduction in the number of alveoli. It results from abnormal maturation of the bronchopulmonary tree [2].
Incidence, Etiology and Classification
The incidence of CCAM is between 1:25,000 and 1:35,000 live births, with males in most series being more commonly affected [2]. The etiology of the entity remains unknown. Hamartomatous change in the terminal bronchioles or an arrest in their embryological development between 7 and 15 weeks of gestation is suspected. Decreased apoptosis also plays a role. The related genes in the pathogenesis are HOXB5, Fgf7, and PDGFB. Exaggerated signaling of the fibroblast growth factor 10 (FGF10) may be responsible for the formation of cystic-like structures during early lung development. FGF10 contributes to lung morphogenesis through its receptor FGFR2, and its signaling is down-regulated. There is increased expression of airway epithelial markers as well as dysregulated expression of genes related to the Ras and several kinases signaling pathways. Also mutations in the DICER1 gene leads to the formation of cystic airways, disruption of branching morphogenesis and mesenchymal expansion, features similar to pleuropulmonary blastoma. There is currently debate about the pathological classification of CTM [1]. Adzick et al. differentiated CCAM to macrocystic and microcystic types.
According to the 2002 Stocker classification, the term congenital pulmonary airway malformation (CPAM), can replace the former CCAM and includes five types:
1. Type 0 (bronchial type, formerly described as acinar dysplasia), bronchial-type airways separated only by abundant mesenchymal tissue.
2. Type 1 (the bronchial/bronchiolar type) and
3. Type 2 (the bronchiolar type) are characterized by cysts >2 cm in diameter and multiple small cysts, respectively.
4. Type 3 CPAM (the bronchiolar/alveolar type): the lesion is solid, and not cystic, because of the excess of bronchiolar structure separated by airspaces that resemble late fetal lung.
5. Type 4 CPAM (the peripheral type) is characterized by peripheral thin-walled, often multiloculated cysts.
CCAM is usually unilateral, mostly left-sided. The significance of bilateral lesions may relate to a genetic predisposition to an underlying cell signalling issue and perhaps increase the likelihood of a predisposition to subsequent malignant change within the CCAM or elsewhere in the lung. Rarely association with aneuploidy is found. Compression of the heart and the vena cava leading to mediastinal shift, polyhydramnios, and fetal hydrops may occur. Moreover, skeletal anomalies, Potter’s syndrome and gastrointestinal atresia may be additional findings [3].
Diagnosis, Differential Diagnosis
CCAM is usually identified during the anatomy ultrasound scanning of the second trimester. Prenatal diagnosis of CCAM has increased due to the advances in antenatal sonography. It can be used to assess size, location, characteristics (macro or microcystic, solid, or mixed lesions), and volume changes with growth, as well as blood supply, mediastinal shift, pleural effusion, or other signs of fetal hydrops. Moreover, the 3D sonographic evaluation allows all cystic lesions within the studied volume to be seen as opaque or echogenic. MRI is another useful tool for imaging CCAM in addition to ultrasound useful both to detect a systemic arterial blood supply and any complications. The differential diagnosis includes bronchopulmonary sequestration, bronchogenic cyst, neurenteric cysts, congenital lobar emphysema, and diaphragmatic hernia [2].
Poor Prognosis and Termination of Pregnancy
Poor prognostic findings are hydrops fetalis, ascites, polyhydramnios, bilateral lung involvement, and a lung-to-thorax transverse area ratio of less than 0.25. The cystic adenomatoid malformation volume ratio which is defined as the estimated volume of the lung lesion divided by head circumference is used to predict prognosis. A ratio >1.6, leads to a poorer prognosis (80% increased risk of fetal hydrops) [1,2]. The increase of the cardiomediastinal shift angle, a novel measure of mediastinal shift, has been significantly associated with an adverse perinatal outcome of CTM. Genetic counseling is necessary and pregnancy termination is an option in cases of poor prognosis. The peak CCAM growth is expected to occur by the 28th gestational week and then the cysts may decrease in size in 20% of the cases. Delivery should be planned in tertiary centres with a neonatal intensive care unit.
Prenatal Interventions
Conservative management, prenatally and postnatally, could be accepted in selected cases [4]. Fetuses with CCAMs and hydrops may be candidates for prenatal intervention where available: cystamniotic shunting, tumor resection before 32 weeks of gestation and percutaneous laser ablation.
Pediatric Surgery
Postnatal assessment is always recommended. Early neonatal surgery is required for symptomatic babies. Elective surgery is prefered in asymptomatic cases in 3–6 months of life to avoid respiratory infections, pneumothorax, or even malignancy. A partial lung resection using an axillary skin crease incision is proposed to obtain a good postoperative quality of life. Although rare entities, the most common relevant malignancies are bronchoalveolar cancer and rhabdomyosarcoma [5]. Usually, a lobectomy in such cases is performed, and then the mortality rates range from 9 to 49%.
Case Report
We present a case of 30 year old pregnant women (G2, para 1), refered to University gynecology and obstetrics clinic in 21 gestational week for 2nd trimester anomaly scan. Her medical and obstetric history were uneventfull. In the 10th gestational week of the current pregnancy she had a Covid 19 infection with mild symptoms. The ultrasound exam showed an adequate fetal growth for gestational age, placental morphology and amniotic fluid index. Fetal lung was markedly abnormal with hyperehogenic and cystic lesions mostly in the right lung pulmonary lobe with displacement of the heart and largest cyst with diameter of 10mm (Figure 1). Both partners were detally counceled and amniocentesis was performed. The quantitative analysis for trisomy 13, 18 and 21 was negative for numeric aberrations. MRI was performed: in the right pulmonary lobe in the hilar area there were 3 cystic changes around the blood vessels, in the parachylar area cyst with diameter of 7-8mm, anterobasal area one cyst with diameter of 10mm and smaller with diameter of 3mm. The findings suggested CCAM, type 2 (Figure 2). After genetic counseling parents decided for pregnancy termination, although they were informed regarding the prognosis and possible interventions.
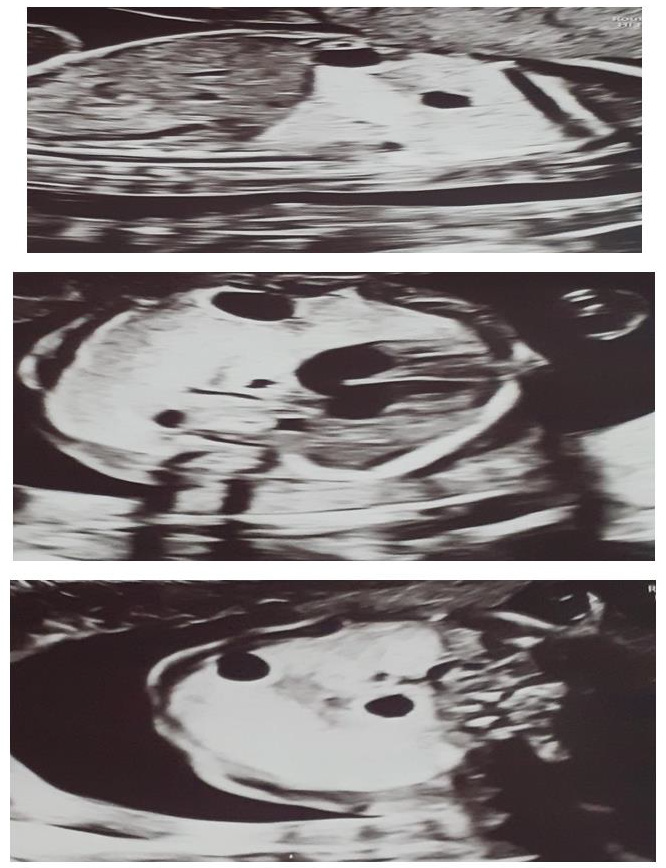
Figure 1: Ultrasound images at the level of the fetal thorax.
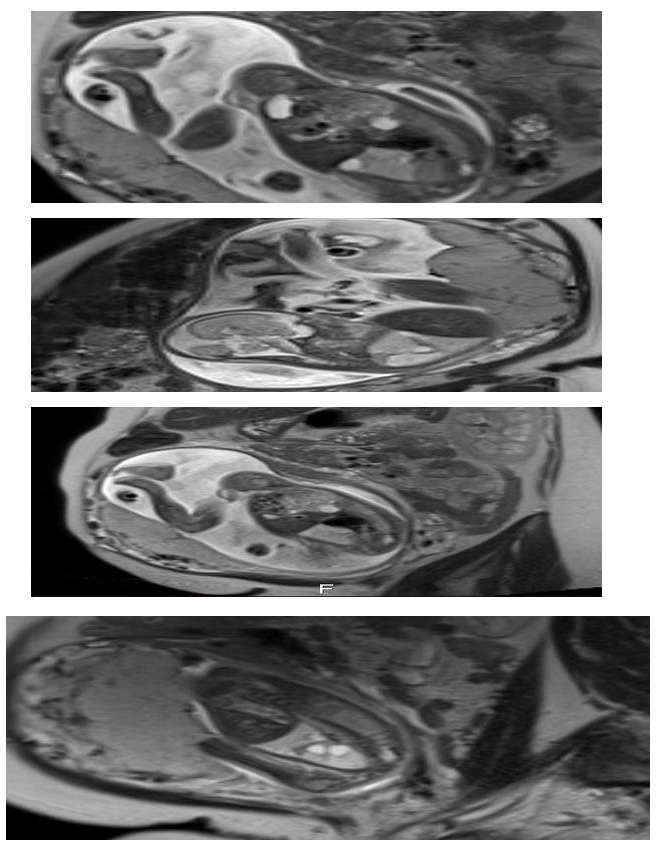
Figure 2: MRI images of the fetus.
Pathohistological examination of the male fetus was in favour of the prenatal diagnosis. In the left pulmonary lobe and upper part of the right pulmonary lobe there were small cysts with a diameter of 0,3-0.6cm, lined with one layer of cubical epithelial cells (CK 7+), and thin fibromuscular layer (SMA+, desmin-). Between cystic changes there were parts of a fibrous tissue. The morphology was in favour of congenital cystic adenoid malformation of the lungs type 2.
Discussion
CCAM is a rare condition usually identified in the second trimester morphology scan. Because of the advances in antenatal sonography prenatal diagnosis of congenital fetal lung disorders has increased. Tran, et al. [5] after a retrospective study of 34 cases with CCAM proposed that asymptomatic neonates should have a postnatal CT even if the CCAM seems to have decreased on antenatal ultrasound. Computerised tomography seems better than X-ray in detecting lesions postnatally. Perinatal mortality of this anomaly can vary from 9% to 49%. In a retrospective study of Shamas, et al. [3] in 14 years period 26 cases were diagnosed around 20 weeks of gestation, all of them unilateral, 31% resolved, 8% decreased, 42% unchanged and 4% increased. Out of them: 16 cases required surgery, 4 were conservatively managed, 12% had associated anomalies. In a study by Mark Davenport, et al. [6] in a 6 years period 67 fetuses had a cystic lung malformation. They were diagnosed in the median age of 21 (19 to 28) gestational weeks, most of them left sided and microcystic, hydrops occured in 7%. Four antenatal interventions were performed: three thoraco-amniotic shunts and one percutaneous intrauterine laser therapy. There was one termination of pregnancy, 2 intrauterine deaths and 96% of the fetuses were born alive. Postnataly 63% had thoracotomy and excisional surgery at an average age of 7.5 months. Two infants (including the case of intrauterine laser therapy) died in the early postnatal period.
In a retrospective cohort study by Chen, et al. [4] in a period of 3 years out of 227 cases prenatally diagnosed as CCAM: 46 pregnancies were terminated, similar number of cases were microcystic and macrocystic. CCAMs presented more offen in male fetuses with polyhydramnios. Thirty one cases received successful surgical resection. In our case although the diagnosis was clear with ultrasound scan, according to most experiences fetal MRI was performed and diagnosis was confirmed. Although we didn’t expect a chromosomical aberration we did an amniocentesis for fetal cariotype. Parents were detally informed by a certified maternalfetal specialist and pediatric surgeons in terms of prognosis, possible outcome and postnatal surgical procedures. In this case CCAM volume ratio (estimated volume of the lung lesion/ head circumference) >1.6 leaded to a poorer prognosis so termination of pregnancy was an option according to the legal procedure in our country.
Conclusion
CCAM is a rare congenital anomaly with reported perinatal mortality as high as 49%. The prenatal and postnatal management remains controversial. Surgical management is the preferred option over conservative management postnatally. Genetic counseling is necessary and termination of pregnancy is an option in cases of poor prognosis [7].
For more Articles on: https://biomedres01.blogspot.com/



No comments:
Post a Comment
Note: Only a member of this blog may post a comment.