Anthrax Toxins and their receptors
Introduction
Under unfavorable growth conditions, B.anthracis undertake
the developmental process of sporulation. B.anthracis spores
are their infectious form, because contact with such spore forms
under favorable conditions can lead to inhalation, skin and
gastrointestinal infections [1]. For example, when spores enter the
lungs, they are phagocytosed by macrophages and dendritic cells.
However, some of them are able to spread throughout the body,
despite the initial immune response. The spores that survive then
transform into vegetative bacilli thanks to the formation of a polyɣ-
D-glutamine capsule and the secretion of anthrax toxin proteins
[2]. The anthrax toxin is composed of two binary combinations
of three soluble proteins: 83 kDa protective antigen (PA83), 90
kDa lethal factor (LF), and 89 kDa edema factor (EF). PA forms
complexes on the surface of host cells. It binds to one of two known
anthrax toxin receptors, tumor endothelial marker-8 (TEM-8) or
capillary morphogenesis protein-2 (CMG-2) [3]. Receptor-bound
PA is proteolytically activated by a cell surface protease to generate
a 63-kDa form (PA63) which oligomerizes, generating ring-shaped
heptameric and octameric pore precursors [4].
These pre-channel oligomers are capable of binding up to three and four LF and/or EF molecules, respectively. The complexes are endocytosed and delivered to an acidic endosomal compartment. The PA oligomer transformed into a translocase channel, allows the transmembrane proton gradient to force lethal factor and edema factor translocation into the cytosol where they carry out their enzymatic functions, (Figure 1) [5]. Bacillus anthracis is a Grampositive, spore-forming, rod-shaped bacterium and is recognized by the presence of the pX01 and pX02 virulence plasmids, which give its unique ability to produce the anthrax toxin [6]. The plasmids pXO1 and pXO2 are very important for the virulence of B. anthracis. The pXO2 plasmid contains genes encoding the poly-D-ɣ-glutamic acid capsule, and the pXO1 plasmid contains genes encoding the toxin components: PA, EF, LF and virulence regulator anthrax toxin activator (AtxA) [7]. AtxA regulates genes encoding anthrax toxins and capsule synthesis [7]. AtxA includes the domains: two helixturn- helix (HTH), which is responsible for binding to DNA, and two phosphotransferase system (PTS) regulation domains (PRDs) and an EIIB-like domain [7]. Due to the presence of the PRDs domain in AtxA, its activity is regulated by phosphorylation strictly dependent on the presence of carbon dioxide [8,9]. These findings have important implications for developing research on the main role anthrax toxins as a major virulence factors at the initial stage of anthrax infection.
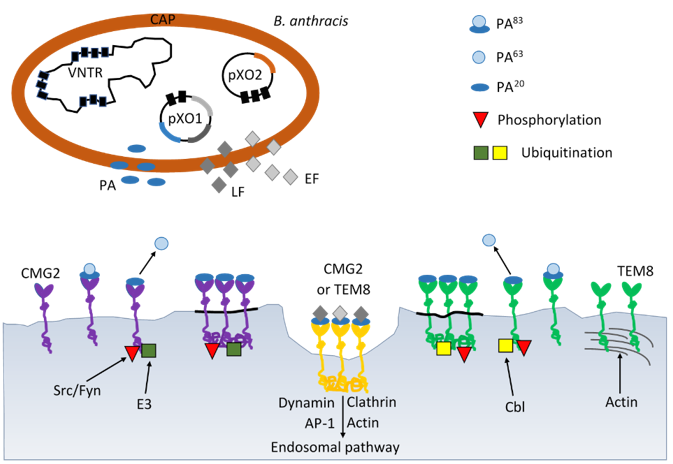
Figure 1: Schematic mechanisms of virulence B. anthracis by anthrax toxin and progression through the endocytic pathway. B. anthracis produces the three subunits of anthrax toxin: protective antigen (PA), lethal factor (LF) and edema factor (EF) encoded on plasmid pXO1 and poly-D-glutamic acid polimer capsule (CAP) ancoded on plasmid pXO2. AP-1 – activator protein 1; Cbl – E3 ubiquitin-protein ligase; CMG2 – capillary morphogenesis gene 2; E3 – ligase; EF – edema factor calmodulindependent adenylate cyclase; Fyn – tyrosine-protein kinase; LF – lethal toxin zinc metalloprotease; Src – Src-like kinase; TEM8 – tumor endothelial marker 8; VNTR – variable number tandem repeat.
Anthrax Toxin
Anthrax toxin is comprised of three nontoxic proteins that combine on eukaryotic host cell surface to form a toxic complex. A tripartite AB-type anthrax toxin is comprised of two catalytic A moieties: lethal factor (LF) and edema factor (EF) and a single receptor-binding B moiety, designated as protective antigen (PA). Lethal factor is a zinc-dependent metalloprotease that, together with protective antigen forms lethal toxin. It is a main virulence factor and the major cause of death for the Bacillus anthracis infected organism [10]. Lethal factor specifically cleaves the N-terminal end of mitogen-activated protein kinase kinases (MAPKKs) (Pellizzari et al. 1999). Because the N-terminal domain of MAPKKs is essential for the interaction between MAPKKs and mitogen-activated protein kinases (MAPKs), the cleavage of this domain impairs the activation of MAPKs [11]. Lethal factor cleavage of MAPKKs leads to the inhibition of three major signaling pathways-ERK1/2 (extracellular signal-regulated kinase), JNK/SAPK (c-Jun N-terminal kinase) and p38 kinases [12].
They are involved in diverse cellular processes including growth, apoptosis, innate and adaptive immune responses and several responses to various forms of cellular stress. According to lethal factor crystal structure, the enzyme comprises four different domains [13]. Domain I is responsible for protective antigen binding. The catalytic site and two zinc-binding motifs are found in C-terminal of domain IV [13]. Several studies have shown that histidine residues play an important role in the catalytic activities of lethal factor. Three histidine residues, His-35, His-42, His-229 are important for lethal factor binding to protective antigen but His- 686, His-690 and specially His-669 are essential for lethal factor catalytic activity [14,15]. Edema factor (EF) is composed of two functional domains, domain I (EFN, residues 1-291) and domain II (residues 292-798) [16]. The first domain (30 kDaN-terminal PA binding domain) interacts with protective antigen whereas the second (43 kDa AC domain and 17 kDa helical domain) interacts with adenylyl cyclase [17].
A series of biochemical studies has been revealed that EF also has two conserved aspartate residues, which coordinate two magnesium ions required for adenylyl cyclase activity [17]. EF is a calmodulin-dependent adenylyl cyclase that increases intracellular cAMP concentration of infected cells. cAMP is a secondary messenger with multiple downstream effectors, including protein kinase A (PKA) and protein activated by cAMP (EPAC). High levels of cAMP generated by ET activate PKA-induced transcriptional changes including modulation of cAMP-responsive element binding (CREB) protein [18]. Study on monocyte-derived cells suggests that CREB and glycogen synthase kinase 3 (GSK-3) are important for the ET-induced expression of anthrax toxin receptor 2 [19]. In addition, cAMP as a second messenger contributes to the regulation of leukocyte chemotaxis and endothelial barrier integrity [20,21]. The results reported by Nguyen, et al. [22] demonstrated that edema toxin (ET) impedes IL-8 driven movement of neutrophils across an endothelium independent of c AMP/PKA activity.
The stability and even the formation of the EF-calmodulin complex depends on the level of calcium bound to calmodulin (CaM) [23]. The binding of calmodulin to EF is a sequential process, first the N-terminal CaM is anchored to the helical domain and next C-terminal CaM region can insert between the catalytic core and helical domains of EF [24]. It leads to a conformational change of C-terminal region to stabilize the catalytic loop of EF for enzymatic activity. According to crystallographic studies residues Leu 667, Ser 668, Arg 671, Arg 672 and Val 694 are implicated in binding of calmodulin to EF [16,25,26]. Makiya, at al. [25] identified these amino acids residues as the binding epitope of EF-neutralizing mAb EF13D, which can neutralize EF in vitro in the subnanomolar range. Other labs have also reported small molecules which inhibit EF by different mechanisms but in the micromolar range [27,28]. Nanomolar affinities are often requested for an efficient competition, which explains that antibody concentration plays a role in toxin neutralization.
A variety of other types of EF inhibitors have been proposed. Especially, various purine and pyrimidine nucleotides with unique preference for the base cytosine were studied [29]. Edema factor and lethal factor forms toxic complexes with protective antigen, edema toxin (ET), which induces tissue swelling and lethal toxin (LT), which can alter cell function and may cause death [4]. The maintenance of homeostasis of the neural microenvironment is responsible for the blood-brain barrier. It is a regulatory interface between the peripheral circulation and the central nervous system (CNS) [30]. The endothelial barrier protects the brain from microorganisms and toxins circulating in the blood. Unfortunately, pathogenic microorganisms have evolved neuroinvasiveness mechanisms to penetrate host cell barriers. In vitro and in vivo studies from several laboratories suggest a principle role for ET in modulating brain endothelial integrity by disrupting the intercellular contacts and a role for LT in promoting penetration of the blood-brain barrier and development of meningitis [31-34].
Edema toxin has been shown to alter host defense like reduced activation of antigen-presenting cells, increased release of cytokines from dendritic cells, impaired chemotaxis and differentiation of T lymphocytes [35]. ET has also been shown to play an important role in the pathogenesis of anthrax-associated shock [36]. Infection with Bacillus anthracis can be cutaneous, gastrointestinal or pulmonary (inhalational). Frequently affected organs include secondary lymph nodes, lung, spleen, kidney, liver, intestinal serosa, heart, and brain proper [37]. Destruction of the organ function is due to the secretion of LT and ET. Some labs have reported that lethal toxin can disrupt endothelial barrier function [37]. The mechanisms causing endothelial dysfunction are stimulation of endothelial apoptosis, alteration of actin fibers and cadherins and mast cell activation [36,37]. Other lab has found that endothelial permeability is under tight control system where hypoxia activates signaling through the Rho-kinase-myosin light chain phosphatase pathway which leads to increased permeability [38].
However, hypoxia can activates p38 MAP kinase signaling leading to heat shock protein 27 (hsp27) phosphorylation which decreases endothelial permeability [39]. The majority studies indicate that anthrax lethal toxin induces the apoptosis of macrophages in an activated caspase-dependent way [40,41]. The cytotoxicity of lethal toxin is related to the activation of the transcription factor- NF-κB and TNF-α (tumor necrosis factoralpha) production in bovine macrophages [42]. It was shown that in bovine macrophages lethal toxin efficiently induces inhibitor-1- κB degradation and enhances the nuclear translocation of NF-κB. Neither protective antigen nor lethal factor alone had any impact of NF-κB activation. Lethal toxin induces apoptosis and necrosis in bone marrow derived macrophages and in activated human peripheral blood monocytes [41,43].
Interestingly, human alveolar macrophages demonstrate significant resistance to all the effects of lethal toxin, including inhibition of cytokine induction, lethal toxin-mediated MEK cleavage and lethal toxin-mediated apoptosis [44]. Lethal toxin, through its effect on the p38 pathway, disrupts glucocorticoid receptor signaling [45]. In vitro study has suggested that lethal toxin may depress murine cardiomyocytes function via an NADPH oxidase-mediated superoxide production mechanism [46]. Series of histological and microbiological studies concerning the effect of LT on intestinal tissues confirm LT-induced intestinal pathology, which is marked by villous blunting, mucosal erosions and ulceration [47-49]. Protective antigen is an 83 kDa pore-forming protein that binds to the anthrax receptors on the surface of the target cells and arrange entry of lethal toxin and edema toxin into the cytosol [50]. Native form PA consists of four domains with different functions: domain 1 - proteolytic activation by furin occurs in it; domain 2 - forms a transmembrane pore to translocate edema factor and lethal factor into the cell and contributes significantly to the receptor interaction; domain 3 - mediates in self-association of nicked form of PA83; domain 4 - primarily involved in binding to anthrax toxin receptor [50,51].
Upon binding to receptors, PA molecules undergo furin cleavage into 20 kDa fragment and 63 kDa subunits that remain cell surface bound. Furin is a critical housekeeping enzyme involved in protoxin activation [52]. Furin is essential for introducing the anthrax toxin into macrophages in highly pathogenic strains. The PA 63 kDa molecule creates a membrane channel that allows the entry of the LF toxin into the cytoplasm of the host cell [53]. It is extremely interesting that the site of cleavage of the PA protein of anthrax toxin has homology with the S1 site of the SARS-CoV2 virus, which is also affected by furin and the transmembrane protease serine 2 (TMPRSS2) [53]. Furthermore, both the anthrax toxin and the SARS-CoV2 virus infect macrophages and respiratory epithelial cells. In both infections, furin, the infected host’s protease, is the initiating enzyme. It is a factor that activates both the anthrax toxin and the SARS-CoV2 virus protein [54]. The characteristic protein sequences affected by furin are common among influenza, measles viruses, flaviviruses and the botulinum toxin [54]. They are the socalled initiation sequences, the presence of which among bacterial or viral strains increases their pathogenicity and virulence. As mentioned earlier, furin is a key host enzyme involved in the activation of protoxins and is therefore an interesting target for the search for its inhibitors. It is highly probable that the tropism and pathogenicity of bacterial strains increases as a result of the action of furin [54,55].
Anthrax Toxin Receptors
Two different cell surface receptors mediate anthrax toxin entry to the cells: ANTXR1, tumor endothelium marker 8 (TEM- 8) and ANTXR2, capillary morphogenesis protein 2 (CMG-2) [1,3]. TEM 8 and CMG 2 are type I membrane proteins containing the domain of von Willebrand factor A, which was originally identified in the blood serum protein as a platelet adhesion factor. ANTXR1 was previously discovered as a tumor endothelium marker, which is present at very low levels in healthy tissues and significantly increased in tumor tissues. ANTXR1 shares many similarities with integrins [56]. The ANTXR1 structural domains are similar to the β1 integrin domains and interact with type I and type VI collagen which also aid in cell migration and extracellular matrix reorganization [57]. On the other hand, the cytoplasmic part of the ANTXR1 receptor, directly anchored to the cytoskeleton of the actin cell, influences cell signal transmission, similar to integrins [58,59].
Cheng at al. for the first time investigated the mechanical signal transduction pathway initiated by the mechanical stimulation of the ANTXR1 receptor and its subsequent conversion to a biological signal in bone marrow stromal cells (BMSCc) [60]. The ANTXR1-initiated mechanotransduction involving the proteins LPR6 and LPR5 (low-density lipoprotein receptor-related protein) partially activates β-catenin to transfer a mechanical signal to the cell nucleus to regulate chondrogenesis [60]. Moreover, further experiments confirmed the interaction of ANTXR1 with actin and fascin actin-bundling protein 1 (FSCN1), which may also suggest the participation of anthrax receptors in the reorganization of the cell cytoskeleton [60]. Both CMG2 and TEM8 receptors have long cytoplasmic domains of 148 and 222 amino acid residues, respectively, like many other signaling receptors, and their physiological roles are related to cell migration and extracellular matrix remodeling [61,62].
Receptors can be post-translationally modified as a result of glycosylation, palmitoylation or ubiquitination [63,64]. Glycosylation affects protein folding in the ER, movement, and function. The TEM8 receptor has putative three glycosylation sites that are necessary for the movement of this protein from the ER and reaching the cell membrane [65]. It was verified that the TEM8 receptor lacking glycosylation did not bind the anthrax toxin in HeLa cells [65]. In contrast, the CMG2 receptor in the same cells, in the absence of glycosylation, could leave the ER and reach the cell membrane where it was able to bind ligand. Both receptors can be ubiquitinated by the action of the host ubiquitin ligase, leading to endocytosis of the clathrin-dependent toxin complex [63]. This process is even necessary for the intracellular activity of the anthrax toxin. S-palmitoylation involves the attachment of a 16-carbon fatty acid to a specific cysteine to form a thioester bond. In proteins, there may be a correlation between palmitoylation and ubiquitination within the same molecule. An example of such a phenomenon is the ubiquitination of the TEM8 receptor, if it has not been palmitoylated before, which leads to its destabilization and premature degradation [66].
The cytoplasmic domain of ANTXR1 and ANTXR2 are important in regulating half-life of the receptors at the plasma membrane [67]. The palmitoylation of cysteine residues increase the half-life of these proteins by preventing its premature clearance for the cell surface [63]. In the cytoplasmic domain, both receptors contain tyrosine residues phosphorylated following binding of protective antigen by receptor which is required for efficient toxin uptake [64]. There are three isoforms of ANTXR1. The ANTXR1-sv1, the longest isoform has 564 amino acids and the medium isoform ANTXR1-sv2 has 386 amino acids [68]; ANTXR1-sv3, the short isoform does not contain the transmembrane domain, so it cannot bind of PA and probably acts as secreted protein [69]. The studies of isoforms have demonstrated that the extracellular and transmembrane domains of these receptors are essential for PA binding, oligomer formation and translocation of anthrax toxin into the cytosol [69].
Toxin Entry into Cells
Toxin entry into host cells begins when protective antigen (PA83) binds to either of two cell surface receptors, ANTXR1 or ANTXR2. Following that PA83 is proteolytically activated by furinlike protease to create an active 63kD-form (PA63). Receptor - bound PA63 has the ability to oligomerize into heptameric or octameric rings, to form a pre-pore that can bind up to three molecules of either edema factor or lethal factor. The toxin-receptor complex is then internalized preferentially via clathrin-mediated endocytosis (Figure 1). This endocytosis appears to be protein depend such as clathrin, dynamin, heterotetrameric adaptor (AP-1) and actin [70]. ANTXR1 and ANTXR2 both could interact with lipoproteinreceptor- related proteins 5 (LRP5) and lipoprotein-receptorrelated proteins 6 (LRP6) [71]. Presumably, there are required for anthrax endocytosis. The large hetero oligomeric complex is then transported to early endosomes where it is incorporated into intraluminal vesicles [69].
The acidic pH of the early endosomes induces structural changes in the PA pre-pore leading to the pore formation as well as to the partial unfolding of edema factor and lethal factor [72]. The current study found that both proteins, EF and LF undergo major conformational changes during binding to PA [69]. These are then translocated through the PA pore across the endosomal membrane. It is understood that the anthrax toxin enzymatic subunits before being released into the cytosol must be transported to late endosomes in a microtubule dependent manner which is essential to protect them from lysosomal proteases. However, many of the details of this sophisticated delivery system remain to be elucidated.
Conclusion
Bacillus anthracis has two virulence factors, a poly-ɣ-Dglutamine
capsule and bipartite toxins. The capsule of B. anthracis
contributes to pathogenesis by blocking phagocytosis. The lethal
toxin (LT) and edema toxin (ET) play a significant role in the
pathogenesis of the disease. The study of the mechanisms by which
these toxins modulate host defense has tremendously improved.
The discovery of anthrax toxin receptors is a relevant for anthrax
pathogenesis. Anthrax toxin receptors can regulate ligand-binding
after conformational changes. A better understanding of anthrax
pathogenesis may allow design of effective inhibitors. Future
studies of anthrax toxins and their receptors promises to yield more
information concerning toxin entry into the cells and therapeutic
applications.
For more Articles on : https://biomedres01.blogspot.com/



No comments:
Post a Comment
Note: Only a member of this blog may post a comment.